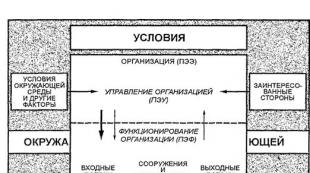
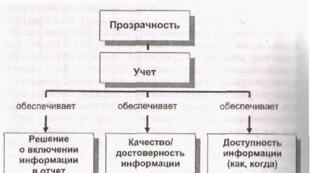
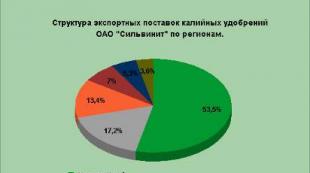
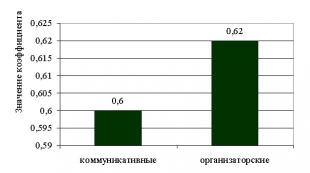
Resistenza chimica dei materiali da costruzione in base alla loro composizione e struttura. Proprietà tecnologiche dei materiali. Resistenza chimica dei materiali Vladislav Aleksandrovich Likhachev x Corso principale di lavoro
Ministero dell'Istruzione Generale e Professionale
Federazione Russa
UNIVERSITÀ STATALE DI INGEGNERIA AMBIENTALE DI MOSCA
A.G.Parshin V.S.Pakhomov D.L.Lebedev
Resistenza chimica dei materiali e protezione dalla corrosione
Laboratorio di laboratorio
A cura del Dr. Tech. Scienze A.A.Shevchenko
Mosca-1998
BBK35.11Χ46
Revisori:
Dipartimento di corrosione, Accademia statale del petrolio e del gas di Mosca. Gubkin;
Dottorato di ricerca tecnologia. Scienze A.S. Abramov, società ambientale "ShanEco", Mosca.
Approvato come sussidio didattico dal consiglio editoriale e editoriale dell'Università statale di ingegneria ambientale di Mosca.
Parshin A.G., Pakhomov V.S., Lebedev D.L.
Χ 46 Resistenza chimica dei materiali e protezione dalla corrosione: laboratorio di laboratorio / Ed. A.A. Shevchenko - M.: MSUIE, 1998.-80 pp.; illustrazione.8.
ISBN 5-230-11142-9
Il workshop contiene quattro lavori di laboratorio sulla corrosione elettrochimica e sulla protezione dei metalli: "L'influenza dell'eterogeneità strutturale sulla corrosione dei metalli durante la riduzione degli ioni idrogeno", "Corrosione dei metalli con depolarizzazione dell'ossigeno", "Potenziali degli elettrodi", "Corrosione da contatto e protezione catodica dei metalli dalla corrosione”. Vengono delineati i concetti di base della teoria della corrosione elettrochimica dei metalli e la metodologia per condurre la ricerca sulla corrosione. Progettato per gli studenti a tempo pieno del 3° e 4° anno che studiano il corso di resistenza chimica dei materiali e protezione dalla corrosione.
ISBN 5-230-11142-9 UDC620.193 BBK 35.11
© A.G.Parshin, V.S.Pakhomov, D.L.Lebedev.1998
© MSUIE, 1998
Prefazione
Il workshop di laboratorio è scritto in conformità con il programma del corso "Resistenza chimica dei materiali e protezione dalla corrosione", previsto nel curriculum di una serie di specialità. Il workshop prevedeva lavori sulla corrosione elettrochimica dei metalli, sviluppati presso il Dipartimento di materiali compositi e protezione dalla corrosione.
Ogni lavoro di laboratorio inizia con una parte teorica. Vengono considerati il meccanismo, la termodinamica e la cinetica della corrosione durante la riduzione catodica degli ioni idrogeno, la depolarizzazione dell'ossigeno, i processi di equilibrio e non equilibrio all'interfaccia metallo-elettrolita e la teoria dei potenziali degli elettrodi.
Quando si presentano questioni teoriche, vengono utilizzati concetti elettrochimici moderni sul meccanismo e sulla cinetica dei processi di corrosione.
L'esecuzione di attività di laboratorio consente agli studenti di comprendere meglio i fondamenti dello studio della corrosione e della protezione dei metalli e infonde anche competenze nella conduzione di ricerche di base sulla corrosione elettrochimica in laboratorio.
Tenere un diario di laboratorio
1) titolo e scopo dell'opera;
2) schema di installazione;
3) una tabella con i risultati degli esperimenti e la loro elaborazione (calcoli, grafici);
4. Conclusioni.
Il diario deve essere tenuto in ordine e immediatamente pulito, ad es. allo stesso modo in cui vengono conservate le registrazioni di tutte le attività di ricerca e sperimentazione.
Lo schema di installazione dovrebbe essere chiaro e comprensibile senza spiegazioni verbali.
È necessario fornire le condizioni sperimentali: materiali testati, superficie dei campioni, composizione e concentrazione degli elettroliti, temperatura, ecc.
I risultati dei test dovrebbero essere registrati in tabelle precompilate, le cui forme sono fornite nella descrizione del lavoro.
Quando si registrano varie quantità, è necessario indicare la loro dimensione. I risultati della misurazione, di norma, sono soggetti a elaborazione aggiuntiva: analitica e grafica (calcolo degli indicatori di massa, volume e profondità di corrosione, potenziali, ecc.). In questi casi è necessario fornire la formula di calcolo e un calcolo completo, ovvero con la sostituzione dei valori sperimentali nella formula e per altri calcoli simili - solo i risultati finali.
Sulla base dei risultati ottenuti ed elaborati di conseguenza, si dovrebbero trarre brevi conclusioni sul lavoro svolto. Al termine, presenta il diario con i dati sperimentali all'insegnante per un visto.
 L'influenza del rame sulla corrosione degli acciai bassolegati Vcor 100% 80% 0,1 0,2 0,3% Cu Esempi di acciai: 10 HSND, 10 G 2 S 1 D, 10 KhDNP, 09 G 2 D, 18 G 2 AF(D)
L'influenza del rame sulla corrosione degli acciai bassolegati Vcor 100% 80% 0,1 0,2 0,3% Cu Esempi di acciai: 10 HSND, 10 G 2 S 1 D, 10 KhDNP, 09 G 2 D, 18 G 2 AF(D)
 Classificazione degli acciai resistenti alla corrosione 1. Gli acciai e le leghe resistenti alla corrosione (inossidabili) sono materiali che resistono alla corrosione elettrochimica negli elettroliti. 2. Il principale elemento legante delle leghe resistenti alla corrosione è il cromo. 3. Il cromo viene introdotto negli acciai inossidabili secondo la regola di Tammann. 4. A seconda degli ambienti in cui vengono utilizzati questi acciai, si distinguono cinque gruppi di acciai e leghe resistenti alla corrosione (inossidabili).
Classificazione degli acciai resistenti alla corrosione 1. Gli acciai e le leghe resistenti alla corrosione (inossidabili) sono materiali che resistono alla corrosione elettrochimica negli elettroliti. 2. Il principale elemento legante delle leghe resistenti alla corrosione è il cromo. 3. Il cromo viene introdotto negli acciai inossidabili secondo la regola di Tammann. 4. A seconda degli ambienti in cui vengono utilizzati questi acciai, si distinguono cinque gruppi di acciai e leghe resistenti alla corrosione (inossidabili).
 Acciai resistenti alla corrosione per ambienti leggermente aggressivi Gli acciai del primo gruppo possono funzionare solo in atmosfera chiusa e corrosione subacquea con asciugatura periodica obbligatoria. In condizioni di atmosfera aperta e di costante corrosione subacquea (soprattutto in acqua calda), nonché di corrosione sotterranea, questi acciai sono soggetti a corrosione per vaiolatura. Questi acciai includono gli acciai al cromo: 08 X 13, 09 X 13, 08 X 17 G (ferritico), 10 X 13, 12 X 13 (martensitico-ferritico), 20 X 13, 30 X 13, 40 X 13 (martensitico). Oltre agli acciai al cromo-manganese e al cromo-nichel con lega economica di nichel (2 -4%) 15 X 17 AG 14, 10 X 14 AG 15, 10 X 14 G 14 N 3 T, 12 X 17 G 14 N 3 , 08 X 18 G 8 N 2 T
Acciai resistenti alla corrosione per ambienti leggermente aggressivi Gli acciai del primo gruppo possono funzionare solo in atmosfera chiusa e corrosione subacquea con asciugatura periodica obbligatoria. In condizioni di atmosfera aperta e di costante corrosione subacquea (soprattutto in acqua calda), nonché di corrosione sotterranea, questi acciai sono soggetti a corrosione per vaiolatura. Questi acciai includono gli acciai al cromo: 08 X 13, 09 X 13, 08 X 17 G (ferritico), 10 X 13, 12 X 13 (martensitico-ferritico), 20 X 13, 30 X 13, 40 X 13 (martensitico). Oltre agli acciai al cromo-manganese e al cromo-nichel con lega economica di nichel (2 -4%) 15 X 17 AG 14, 10 X 14 AG 15, 10 X 14 G 14 N 3 T, 12 X 17 G 14 N 3 , 08 X 18 G 8 N 2 T
 Acciai resistenti alla corrosione (inossidabili) per ambienti salini Il secondo gruppo di acciai resistenti alla corrosione (inossidabili) viene utilizzato in ambienti salini a basse temperature, in particolare durante la corrosione marina. Una maggiore resistenza alla corrosione si ottiene mediante un'ulteriore lega economica di acciai al Ni (5 – 8%). Esempi: 09 X 15 N 8 Yu, 07 X 16 N 6, 08 X 17 N 5 M 3 (l'acciaio è utilizzato in ambienti con acido solforico), 09 X 17 N 7 Yu 1 (gli acciai sono utilizzati in condizioni di corrosione marina).
Acciai resistenti alla corrosione (inossidabili) per ambienti salini Il secondo gruppo di acciai resistenti alla corrosione (inossidabili) viene utilizzato in ambienti salini a basse temperature, in particolare durante la corrosione marina. Una maggiore resistenza alla corrosione si ottiene mediante un'ulteriore lega economica di acciai al Ni (5 – 8%). Esempi: 09 X 15 N 8 Yu, 07 X 16 N 6, 08 X 17 N 5 M 3 (l'acciaio è utilizzato in ambienti con acido solforico), 09 X 17 N 7 Yu 1 (gli acciai sono utilizzati in condizioni di corrosione marina).
 Acciai per ambienti a media corrosività Per ambienti a media corrosività si intendono soluzioni di sali a diverse temperature, nonché soluzioni deboli di alcuni acidi. Gli acciai del terzo gruppo sono gli acciai inossidabili più comuni con ampia applicazione. Tra questi acciai possiamo distinguere: a) acciai - sostitutivi degli acciai ad alto contenuto di nichel: 15 X 25 T, 15 X 28, 08 X 22 N 6 T, 12 X 21 N 5 T. b) acciai con un rapporto ottimale di cromo-nichel rapporto (Cr: Ni = 18: 9, 18: 10): 12 X 18 N 9 T e 12 X 18 N 10 T, 17 X 18 N 9, 12 X 18 N 10 B, 08 X 18 N 10, 12 X 18 N 12 T, 08 X 18 N 12 B, 06 X 18 N 11, ecc.
Acciai per ambienti a media corrosività Per ambienti a media corrosività si intendono soluzioni di sali a diverse temperature, nonché soluzioni deboli di alcuni acidi. Gli acciai del terzo gruppo sono gli acciai inossidabili più comuni con ampia applicazione. Tra questi acciai possiamo distinguere: a) acciai - sostitutivi degli acciai ad alto contenuto di nichel: 15 X 25 T, 15 X 28, 08 X 22 N 6 T, 12 X 21 N 5 T. b) acciai con un rapporto ottimale di cromo-nichel rapporto (Cr: Ni = 18: 9, 18: 10): 12 X 18 N 9 T e 12 X 18 N 10 T, 17 X 18 N 9, 12 X 18 N 10 B, 08 X 18 N 10, 12 X 18 N 12 T, 08 X 18 N 12 B, 06 X 18 N 11, ecc.
 Acciai per l'uso in ambienti con maggiore corrosività Questi tipi di acciai sono stati sviluppati per aumentare la resistenza chimica nelle soluzioni calde di Na. Cl e in soluzioni acide. Per aumentare la resistenza degli acciai, viene utilizzata un'ulteriore lega con molibdeno e rame, e negli acciai di questo gruppo spesso si sforzano di mantenere una struttura austenitica, conveniente in termini tecnologici, che richiede un'ulteriore lega di acciai con nichel. A causa dell'elevato contenuto di componenti leganti, principalmente nichel, gli acciai di questo gruppo sono piuttosto costosi. Esempi di acciai del gruppo sono: 10 X 17 N 13 M 2 T 08 X 17 N 13 M 3 T, 08 X 17 N 15 M 3 T, 04 X 28 MDT, 03 X 28 MDT, 06 X 28 MT.
Acciai per l'uso in ambienti con maggiore corrosività Questi tipi di acciai sono stati sviluppati per aumentare la resistenza chimica nelle soluzioni calde di Na. Cl e in soluzioni acide. Per aumentare la resistenza degli acciai, viene utilizzata un'ulteriore lega con molibdeno e rame, e negli acciai di questo gruppo spesso si sforzano di mantenere una struttura austenitica, conveniente in termini tecnologici, che richiede un'ulteriore lega di acciai con nichel. A causa dell'elevato contenuto di componenti leganti, principalmente nichel, gli acciai di questo gruppo sono piuttosto costosi. Esempi di acciai del gruppo sono: 10 X 17 N 13 M 2 T 08 X 17 N 13 M 3 T, 08 X 17 N 15 M 3 T, 04 X 28 MDT, 03 X 28 MDT, 06 X 28 MT.
 Leghe a base di nichel per ambienti molto aggressivi Per fluidi con elevata aggressività si intendono soluzioni calde di acido solforico e cloridrico. In ambienti così aggressivi i materiali metallici più resistenti sono le leghe a base di nichel. Ad esempio, la lega KhN 65 MV è stabile a temperature elevate in ambienti di acido solforico e cloridrico, in acido acetico concentrato. La lega N 70 MF è consigliata per l'uso in soluzioni di acido solforico e cloridrico; la lega è più resistente alla corrosione intergranulare.
Leghe a base di nichel per ambienti molto aggressivi Per fluidi con elevata aggressività si intendono soluzioni calde di acido solforico e cloridrico. In ambienti così aggressivi i materiali metallici più resistenti sono le leghe a base di nichel. Ad esempio, la lega KhN 65 MV è stabile a temperature elevate in ambienti di acido solforico e cloridrico, in acido acetico concentrato. La lega N 70 MF è consigliata per l'uso in soluzioni di acido solforico e cloridrico; la lega è più resistente alla corrosione intergranulare.
 Aumento della densità del calcestruzzo 4. Introduzione di additivi polimerici 4. 1. introduzione di una piccola quantità di additivi polimerici dallo 0,2 al 3% nella miscela di calcestruzzo (lattici, resine polimeriche); 4. 2. produzione di calcestruzzo a base di legante polimerico (soluzioni polimeriche e calcestruzzo polimerico); Fornito come miscela secca e indurente in barattoli. 4. 3. impregnazione di manufatti in calcestruzzo preconfezionato e in cemento armato con composti polimerici o monomeri con successiva polimerizzazione direttamente nel corpo del calcestruzzo (polimeri del calcestruzzo); 4. 4. rinforzo del calcestruzzo con fibre polimeriche (produzione di calcestruzzo fibrorinforzato)
Aumento della densità del calcestruzzo 4. Introduzione di additivi polimerici 4. 1. introduzione di una piccola quantità di additivi polimerici dallo 0,2 al 3% nella miscela di calcestruzzo (lattici, resine polimeriche); 4. 2. produzione di calcestruzzo a base di legante polimerico (soluzioni polimeriche e calcestruzzo polimerico); Fornito come miscela secca e indurente in barattoli. 4. 3. impregnazione di manufatti in calcestruzzo preconfezionato e in cemento armato con composti polimerici o monomeri con successiva polimerizzazione direttamente nel corpo del calcestruzzo (polimeri del calcestruzzo); 4. 4. rinforzo del calcestruzzo con fibre polimeriche (produzione di calcestruzzo fibrorinforzato)

 Modulo 7. Metodi di protezione dei metalli dalla corrosione elettrochimica. Lezione 7. 3 PROTEZIONE ELETTROCHIMICA DEI METALLI DALLA CORROSIONE La protezione catodica consiste nello spostare il potenziale del metallo di una struttura corrosiva sul lato negativo collegandolo al polo negativo della sorgente di corrente.
Modulo 7. Metodi di protezione dei metalli dalla corrosione elettrochimica. Lezione 7. 3 PROTEZIONE ELETTROCHIMICA DEI METALLI DALLA CORROSIONE La protezione catodica consiste nello spostare il potenziale del metallo di una struttura corrosiva sul lato negativo collegandolo al polo negativo della sorgente di corrente.
 Modulo 7. Metodi di protezione dei metalli dalla corrosione elettrochimica. Lezione 7. 3 Schema di corrosione della protezione catodica
Modulo 7. Metodi di protezione dei metalli dalla corrosione elettrochimica. Lezione 7. 3 Schema di corrosione della protezione catodica
 Modulo 7. Metodi di protezione dei metalli dalla corrosione elettrochimica. Lezione 7.2 La protezione protettiva si basa sulle caratteristiche di corrosione di due metalli a contatto. Secondo la teoria della corrosione da contatto, quando un metallo positivo M 2 entra in contatto con un metallo più negativo M 1, il potenziale del metallo M 2 si sposta sul lato negativo e la sua corrosione diminuisce o si arresta completamente.
Modulo 7. Metodi di protezione dei metalli dalla corrosione elettrochimica. Lezione 7.2 La protezione protettiva si basa sulle caratteristiche di corrosione di due metalli a contatto. Secondo la teoria della corrosione da contatto, quando un metallo positivo M 2 entra in contatto con un metallo più negativo M 1, il potenziale del metallo M 2 si sposta sul lato negativo e la sua corrosione diminuisce o si arresta completamente.
 Modulo 7. Metodi di protezione dei metalli dalla corrosione elettrochimica. Lezione 7.3 La protezione anodica viene utilizzata solo per i metalli soggetti a passivazione in un ambiente corrosivo. Si tratta di spostare il potenziale del metallo dalla regione di dissoluzione attiva alla regione di passivazione utilizzando una fonte di corrente esterna.
Modulo 7. Metodi di protezione dei metalli dalla corrosione elettrochimica. Lezione 7.3 La protezione anodica viene utilizzata solo per i metalli soggetti a passivazione in un ambiente corrosivo. Si tratta di spostare il potenziale del metallo dalla regione di dissoluzione attiva alla regione di passivazione utilizzando una fonte di corrente esterna.
 Modulo 7. Metodi di protezione dei metalli dalla corrosione elettrochimica. Lezione 7. 3 Diagramma di corrosione della protezione anodica
Modulo 7. Metodi di protezione dei metalli dalla corrosione elettrochimica. Lezione 7. 3 Diagramma di corrosione della protezione anodica
MINISTERO DELL'ISTRUZIONE E DELLA SCIENZA DELLA FEDERAZIONE RUSSA ISTITUTO EDUCATIVO DELLO STATO FEDERALE DI ISTRUZIONE PROFESSIONALE SUPERIORE "UNIVERSITÀ STATALE TECNOLOGICA DEI POLIMERI VEGETALI DI SAN PIETROBURGO" T. L. Lukanina, I. Mikhailova, M. A. Radin RESISTENZA CHIMICA DEI MATERIALI E PROTEZIONE CONTRO LA CORROSIONE Libro di testo San Pietroburgo 2014 1 UDC 546(075) BBK 24.1я7 L 840 Lukanina T. L., Mikhailova I. S., Radin M. A. Resistenza chimica dei materiali e protezione dalla corrosione: libro di testo. manuale.− San Pietroburgo: SPbGTURP, 2014. – 85 p. Il manuale fornisce idee moderne sulla termodinamica e sulla cinetica della corrosione chimica, sui meccanismi e sui vari tipi di corrosione elettrochimica, tenendo conto degli ambienti e delle condizioni caratteristici delle apparecchiature dell'industria chimica. Vengono delineati i principi scientifici per la creazione di acciai e leghe resistenti alla corrosione e i metodi per proteggere i materiali strutturali dalla corrosione. Il libro di testo è offerto agli studenti delle università tecniche e delle facoltà che studiano specialità tecnologiche. Può essere utile per studenti laureati, lavoratori scientifici e ingegneristici di imprese chimiche, organizzazioni di progettazione e ricerca coinvolte nella protezione dei metalli dalla corrosione. Revisori: Dr. Chem. scienze, prof. Dipartimento di Chimica ITMO dello Stato di San Pietroburgo. facoltà Slobodov A. A.; Dottorato di ricerca chimico. Scienze, professore associato Dipartimento di Chimica Organica, Università Tecnica Statale del Turkmenistan di San Pietroburgo, Evdokimov A. N. Consigliato per la pubblicazione dal Consiglio editoriale e editoriale dell'Università come supporto didattico. Redattore e correttore di bozze T. A. Smirnova Tech. Redattore L.Ya Titova Templan 2014, pos. 45 ______________________________________________________________ Presentato per la pubblicazione il 14/04/14 Formato 60×84/16. Tipo di carta n. 1. Stampa offset. Stampa il foglio 5.5. Uch.-ed. fogli 5,5. Tiratura 100 copie. Edizione n. 45. Prezzo "C". Ordine Risograph dell'Università tecnologica statale dei polimeri vegetali, San Pietroburgo, 198095, st. Ivana Chernykh, 4 Università tecnologica statale dei polimeri vegetali di San Pietroburgo, 2014 Lukanina T. L., Mikhailova I. S., Radin M. A., 2014 2 INTRODUZIONE LA CORROSIONE dei materiali è la distruzione spontanea dovuta all'interazione chimica o elettrochimica con l'ambiente. La distruzione di metalli e leghe dovuta alla corrosione differisce da altri tipi di distruzione spontanea, come l'erosione o l'usura, che sono causati dall'interazione meccanica con un corpo o con l'ambiente. Per la maggior parte dei metalli e delle leghe, in condizioni operative, lo stato ossidato (ionico) è più stabile, al quale passano a causa della corrosione. Il termine "corrosione" deriva dal latino "corrodere", che significa "corrodere". Le perdite dovute alla corrosione sono solitamente suddivise in dirette e indirette. LE PERDITE DIRETTE comprendono: − il costo del metallo che si trasforma in prodotti di corrosione, nonché il costo delle apparecchiature corrose che si guastano prima che sia trascorso il periodo di ammortamento previsto dalle condizioni tecniche. Inoltre, di norma, il costo delle attrezzature di produzione supera di gran lunga il costo del metallo con cui sono realizzate; − un aumento delle tolleranze per la corrosione, che porta non solo ad un aumento del consumo di metallo, ma anche al deterioramento della progettazione. Ad esempio, per una tubazione con lo stesso diametro esterno, una maggiore tolleranza di metallo porta ad una diminuzione della sezione trasversale utile e, di conseguenza, ad una diminuzione della portata del tubo; − costi per misure di protezione sotto forma di rivestimenti metallici e non metallici, materiali resistenti alla corrosione, inibitori, ecc. I DANNI INDIRETTI dovuti alla corrosione sono causati dai seguenti motivi: − costi aggiuntivi per riparazioni non programmate di apparecchiature guastate; − sottoproduzione a seguito di fermate per riparazioni di apparecchiature danneggiate dalla corrosione; − la possibilità di incidenti, esplosioni e incendi; − fuoriuscita di prodotti dovuta alla formazione di fori passanti nelle pareti delle apparecchiature a seguito di corrosione; 3 – riduzione della sezione trasversale delle tubazioni a seguito della deposizione di prodotti di corrosione, che richiede ulteriore consumo di energia per mantenere la potenza richiesta del flusso del fluido; − riduzione della qualità del prodotto a causa della contaminazione con prodotti della corrosione. La scienza della corrosione e della protezione dei metalli studia l'interazione di metalli e leghe con un ambiente corrosivo, stabilisce il meccanismo di questa interazione e i suoi principi generali. La base scientifica per lo studio della corrosione e della protezione dei metalli è la scienza dei metalli e la chimica fisica. L'obiettivo pratico finale di questa scienza è proteggere dalla distruzione corrosiva dei metalli durante la lavorazione e il funzionamento delle apparecchiature. Capitolo 1. INFORMAZIONI GENERALI SULLA CORROSIONE 1.1. Termodinamica e cinetica dei processi di corrosione La causa principale della corrosione è l'instabilità termodinamica dei metalli in vari ambienti in determinate condizioni esterne. La possibilità fondamentale del verificarsi spontaneo del processo di corrosione è determinata dalle leggi della termodinamica. Il verificarsi di qualsiasi processo spontaneo è accompagnato da una diminuzione dell'energia libera. La possibilità del verificarsi spontaneo di processi di corrosione può essere valutata mediante variazioni del potenziale isobarico-isotermico ΔG1. Un processo spontaneo di ossidazione del metallo - corrosione - è possibile se ∆G< 0, то есть знак ―минус‖ означает, что изобарно−изотермический потенциал убывает и свободная энергия системы уменьшается. Наоборот, если ∆G > 0, allora il segno più per il potenziale isobarico-isotermico corrisponde ad un aumento dell'energia libera, che indica l'impossibilità del verificarsi spontaneo di questa reazione. POTENZIALE ISOBARIO-ISOTERMICO (ΔG) – Energia di Gibbs, uno dei potenziali termodinamici, pari a ΔG = ΔН - TΔS, dove ΔН, ΔS, Т - entalpia, entropia, temperatura termodinamica del sistema. ΔG è proporzionale al numero di particelle nel sistema. Definito come una particella, è chiamato potenziale chimico. ΔG è conveniente per descrivere processi in cui è possibile lo scambio tra la materia e i corpi circostanti. In un processo di equilibrio isotermo che avviene a pressione costante, questo è il lavoro totale del sistema meno il lavoro contro le forze di pressione esterne. È pari alla diminuzione del potenziale isobarico-isotermico (cioè del massimo lavoro utile). 1 4 Tuttavia, sebbene la termodinamica permetta di determinare quanto il sistema studiato è lontano dallo stato di equilibrio ed è capace di una reazione spontanea, ciò non indica la velocità di questo processo. La velocità di corrosione è determinata dalla sua cinetica. Una caratteristica dei processi di corrosione è la loro natura eterogenea. Ciò è dovuto al fatto che la distruzione del metallo avviene all'interfaccia tra due fasi che hanno diversi stati di aggregazione, ad esempio all'interfaccia metallo-liquido, metallo-gas. In tali condizioni, il fattore determinante per la cinetica dell'interazione chimica è la velocità dell'interazione chimica, oppure la fornitura di reagenti dai volumi delle fasi in contatto all'interfaccia dove avviene l'interazione della corrosione e la rimozione dei prodotti di reazione nella direzione opposta direzione. Nel primo caso, la velocità di corrosione è determinata dal controllo cinetico, nel secondo dal controllo della diffusione. In alcuni casi, la velocità di corrosione è determinata dal controllo misto diffusione-cinetica. Per un processo stazionario, la velocità della reazione chimica e la velocità di diffusione sono uguali. Il processo di corrosione è costituito da diverse fasi, che possono avvenire in sequenza o in parallelo. La velocità totale allo stato stazionario di un processo di corrosione costituito da più fasi successive è determinata dalla velocità della fase più lenta. Quando le singole fasi del processo si verificano in parallelo, la velocità di corrosione totale è determinata dalla fase più veloce. Pertanto, se una delle fasi procede molto più velocemente dell’altra, la seconda fase può essere trascurata. 1.2. Classificazione dei processi di corrosione in base al meccanismo di insorgenza Indipendentemente dall'enorme varietà di condizioni per il verificarsi dei processi di corrosione, questi possono essere classificati in base a due caratteristiche principali: - in base al meccanismo del processo - in base alla natura della distruzione della corrosione . Esistono corrosione CHIMICA ed ELETTROCHIMICA. 5 si verifica nei gas secchi (corrosione del gas) e nei liquidi non elettroliti2 (corrosione dei non elettroliti). Se il mezzo non è un elettrolita (gas secchi o liquidi non elettroliti), lo scambio di elettroni avviene direttamente tra il metallo riducente (rosso) e qualsiasi sostanza contenente un elemento elettronegativo in grado di attrarre a sé gli elettroni - l'agente ossidante (bue ), ad esempio: Fe + Сl2 → FeCl2 (1) CORROSIONE CHIMICA rosso Bilancio del trasferimento di elettroni ox Feº − 2ē → Fe2+ 1 Сl2º + 2ē → 2Cl− Feº + 2Сlº = Fe2+ + 2Cl− LA CORROSIONE ELETTROCHIMICA è il processo di interazione di un metallo con un mezzo corrosivo (soluzione elettrolitica), in cui La ionizzazione degli atomi metallici e la riduzione della componente ossidante dell'ambiente corrosivo non avvengono in un unico atto e le loro velocità dipendono dal potenziale dell'elettrodo. Le caratteristiche distintive del processo di corrosione elettrochimica sono le seguenti: – verificarsi simultaneo di due processi separati: ossidativo (dissoluzione del metallo) e riducente (sviluppo di idrogeno, riduzione di ossigeno, rilascio di metallo dalla soluzione, ecc.); – il processo di dissoluzione del metallo è accompagnato dal movimento direzionale degli elettroni nel metallo e degli ioni nell’elettrolita, cioè la presenza di corrente elettrica; – i prodotti della corrosione si formano a seguito di reazioni secondarie. I processi redox che si verificano durante la corrosione elettrochimica possono essere presentati sotto forma delle seguenti reazioni: Ме − nē→ Меn+ (processo anodico) (2) n− R(ox) + nē → R (rosso) (processo catodico), (3 ) dove R(ox) è un agente ossidante; I NON ELETTROLITI sono sostanze le cui soluzioni acquose e fusioni non conducono corrente elettrica. Contengono legami covalenti non polari o basso-polari che non si scompongono in ioni. Gas, solidi (non metalli) e composti organici (saccarosio, benzina, alcol) non conducono corrente elettrica. 2 6 R n−(rosso) – forma ridotta dell'agente ossidante; nē – numero di elettroni trasferiti. Un esempio di corrosione elettrochimica è il processo di ossidazione (arrugginimento) del ferro sotto l'influenza dell'acqua: Fe − 2ē → Fe2+ (processo anodico - dissoluzione del ferro) H2O + ½О2 +2ē → 2OH– (processo catodico - riduzione dell'ossigeno) −−−−−− −−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−− − Fe 2+ + 2OH – → Fe(OH)2 (formazione di prodotti di corrosione) Le reazioni (2) e (3) procedono coniugatamente, ma obbediscono alle proprie leggi cinetiche. In questo caso è necessario rispettare le condizioni di stazionarietà del processo, ovvero uguaglianza dei tassi di ossidazione del metallo e riduzione dell'agente ossidante. Queste reazioni possono essere geograficamente separate e avvenire su diverse parti della superficie. Dalle condizioni di stazionarietà ne consegue che è sufficiente rallentare una delle reazioni coniugate affinché la velocità dell'intero processo diminuisca. 1.3. Classificazione dei processi di corrosione in base alla natura della distruzione per corrosione In base alla natura della distruzione per corrosione del metallo, si distinguono CORROSIONE GENERALE E LOCALE. LA CORROSIONE GENERALE si estende su tutta la superficie del metallo; può essere uniforme o irregolare (Fig. 1. - 1.2). 1 2 4 5 7 8 3 6 9 Fig. 1. Tipi di danni da corrosione In molti casi di corrosione si tratta di corrosione generale, quando ad esempio, a causa dell'esposizione del metallo agli elettroliti, i prodotti della corrosione non rimangono sulla superficie metallica. Pertanto, durante l'interazione del rame con acido nitrico, del ferro con acido cloridrico, dell'alluminio con alcali caustici, dello zinco con acido solforico, ecc., si osserva un'intensa corrosione generale. La corrosione generale uniforme è uno dei tipi di corrosione meno pericolosi, a condizione che il il tasso di dissoluzione del metallo dovuto al danno da corrosione non supera gli standard determinati dai requisiti per il funzionamento dell'apparecchio, del dispositivo, ecc. Se il metallo è sufficientemente spesso, la corrosione ha scarso effetto sulla riduzione della resistenza meccanica della struttura sotto sollecitazioni uniformemente distribuite sulla sezione trasversale (tensione, compressione). Tuttavia, la corrosione generale può essere pericolosa quando le parti sono sottoposte a flessione e torsione, poiché in questo caso vengono distrutti gli strati più caricati. CORROSIONE LOCALE - provoca la distruzione solo di alcune aree della superficie metallica, mentre il resto non viene distrutto o il tasso di distruzione è molto basso. Le manifestazioni della corrosione locale sono molto diverse e possono avere diversi gradi di disuniformità, pertanto la corrosione locale si divide in CORROSIONE PER MACCHIE, ULCERE, POTING, CORROSIONE SELETTIVA, CORROSIONE, CRACKING CORROSIVO. INTERCRYSTALLITE Corrosione per macchie, ulcere, punti: i tipi di corrosione locale differiscono nel rapporto tra il diametro dell'area distrutta e la sua profondità (Fig. 1., 4–6). Con la corrosione spot, il diametro dell'area distrutta è molto maggiore della sua profondità, con la distruzione ulcerosa, il diametro dell'area distrutta è commisurato alla sua profondità, e con la corrosione per vaiolatura, il diametro dell'area distrutta è molto inferiore alla sua profondità . In alcuni casi la corrosione per vaiolatura può provocare la distruzione, il che è particolarmente pericoloso per tubazioni, serbatoi, ecc. La corrosione per vaiolatura è tipica dei metalli suscettibili di passivazione - frenatura (cromo, alluminio, acciai inossidabili, ecc.). Lo stato passivo dei metalli può essere interrotto in alcune zone più deboli della superficie, ad esempio nei pori di una pellicola protettiva, in luoghi di inclusioni intermetalliche (ossidi, scorie, ecc.). CORROSIONE SELETTIVA – si divide in: selettiva per componente (selettiva) o selettiva per struttura (intercristallina). 8 Un esempio di CORROSIONE SELETTIVA DEI COMPONENTI è la dezincificazione dell'ottone. La dezincificazione dell'ottone3 consiste nel fatto che lo zinco penetra nell'elettrolita (di solito a pH neutro o leggermente acido) più intensamente del rame. Sulla superficie dell'ottone si forma uno strato sciolto di rame, che contribuisce ad aumentare la corrosione elettrochimica (l'aspetto di una cella microgalvanica). Un esempio di corrosione STRUTTURALE-SELETTIVA è la corrosione della ghisa grigia. La corrosione strutturale selettiva della ghisa grigia consiste nella distruzione predominante della componente ferritica, con conseguente formazione di uno scheletro di grafite riempito di prodotti di corrosione (Fig. 1−3). Allo stesso tempo, la resistenza meccanica della ghisa diminuisce drasticamente. La CORROSIONE INTERCRISTALLITE è caratterizzata dalla distruzione selettiva del metallo lungo i bordi dei grani (Fig. 1–7). Di conseguenza, con un piccolo cambiamento nella massa e nell'aspetto, può verificarsi una perdita molto significativa delle proprietà di resistenza del metallo (Fig. 2). 40fig. 2. Perdita delle proprietà di resistenza del metallo durante vari tipi di distruzione per corrosione 1 – corrosione uniforme; 2 – corrosione intergranulare 20 Perdita di w, % 60 2 0 1 2 4 Perdita di peso, kg/cm2 In casi estremi, con la completa distruzione dei bordi dei grani a causa della corrosione intergranulare, la lega può sgretolarsi in polvere. LA CORROSIONE CRACKING è un caso di corrosione locale in cui la distruzione avviene nella direzione perpendicolare alla massima sollecitazione a trazione (Fig. 1 - 8). 3 Ottone – lega rame-zinco. 9 È tipico che in questo caso una cricca da corrosione possa propagarsi lungo i bordi dei grani o tagliare il corpo cristallino. La distruzione per fatica da corrosione (con esposizione simultanea ad un ambiente corrosivo e carichi alternati o ciclici) procede in modo simile a quella durante la fessurazione per corrosione. LA CORROSIONE DEL SOTTOSUPERFICIE, partendo dalla superficie, si diffonde prevalentemente sotto la superficie, il che spesso porta alla delaminazione e al rigonfiamento del metallo (Fig. 1− 9), ad esempio alla formazione di bolle su lamiere di bassa qualità durante il processo di incisione o corrosione durante il funzionamento di recipienti e apparecchi. 1.4. Indicatori di corrosione Per quantificare la velocità di corrosione si utilizzano indicatori di corrosione: MASSOMETRICI, VOLUMETRICI, DI PROFONDITÀ, MECCANICI, CORRENTE. 2 L'indice di corrosione MASSOMETRICO (Km, g/m ∙h) è la variazione della massa del metallo come risultato della corrosione per unità di superficie (s) del campione e per unità di tempo. La variazione della massa di un campione viene solitamente determinata come la differenza tra la massa del campione prima del test (mо) e la sua massa dopo il test (m1) nel tempo (τ) con la rimozione dei prodotti della corrosione (perdita di massa metallica) . K m = (m0 − m1)/s ∙ τ . (4) 3 2 L'indice di corrosione VOLUMETRICO (Kν, cm /m h) è il volume (Vo) di gas assorbito o rilasciato durante il processo di corrosione, per unità di superficie metallica e per unità di tempo K m = V0 /s ∙ τ. (5) L'indice di corrosione PROFONDA (P, mm/anno) tiene conto della riduzione dello spessore del metallo dovuta alla corrosione, espressa in unità lineari e per unità di tempo. Questo indicatore è utile quando si confronta la corrosione di metalli con densità diverse. Il passaggio dall'indicatore massometrico a quello di profondità può essere effettuato, nel caso di corrosione uniforme, utilizzando la formula P = K m ∙ 8,76 /γ, (6) dove γ è la densità del metallo. L'indicatore di corrosione MECCANICA è determinato dalla variazione di uno dei principali indicatori delle proprietà meccaniche di un metallo durante un certo tempo del processo di corrosione ed è espresso in unità relative o in percentuale. Se la resistenza alla trazione viene utilizzata come indicatore meccanico, la variazione dell'indicatore di resistenza (Kσ) è determinata dalla formula Kσ = Δσb/σb, (7) dove σb è la resistenza alla trazione prima della corrosione, ∆σb è il variazione della resistenza alla trazione del metallo dopo la corrosione nel tempo τ. L'indicatore di corrosione corrente consente, utilizzando la formula di Faraday, di calcolare la quantità di metallo corroso in base all'entità della corrente di corrosione Km = IA /nF, (8) dove Km è la velocità di corrosione in base alla variazione nella massa metallica; I – forza attuale; F – costante di Faraday; n – valenza metallica; A è la massa atomica del metallo. Quando si valuta qualitativamente e quantitativamente la resistenza alla corrosione dei metalli, si consiglia di utilizzare una scala a dieci punti4. Va notato che, sebbene questa scala per valutare la resistenza alla corrosione dei metalli sia diventata più diffusa, soprattutto nell'industria chimica, non è universale, poiché per apparecchiature per scopi diversi, i tassi di corrosione consentiti possono differire di un ordine di grandezza o Di più. Tabella 1 Dieci -Scala ballica della resistenza alla corrosione di metalli e leghe Gruppo di resistenza completamente persistente, molto persistente resistente bassa resistente instabile velocità di corrosione, mm/anno inferiore a 0,001 oltre 0,001 fino a 0,005 fino a 0,01 fino a 0,01 fino a 0,05 oltre 0,05 oltre 05. a 0,1 oltre 0,1 a 0,5 oltre 0,5 a 1,0 oltre 1,0 a 5,0 oltre 5,0 a 10,0 oltre 10,0 PUNTEGGIO 1 2 3 4 5 6 7 8 9 10 Sistema unificato di protezione contro la corrosione e l'invecchiamento. Strutture sotterranee. Requisiti generali per la protezione dalla corrosione. GOST 9.602−2005, http://www.gosthelp.ru/gost/gost206.html 4 11 Capitolo 2. CORROSIONE CHIMICA DEI METALLI La corrosione chimica dei metalli è un processo spontaneo della loro distruzione dovuto all'interazione chimica con l'ambiente esterno. Le caratteristiche distintive del processo di corrosione chimica sono le seguenti: – implementazione dei processi di ossidazione e riduzione in un'unica fase; – assenza di corrente elettrica; – la formazione di prodotti di corrosione direttamente sulla superficie dove avviene la sua distruzione. LA CORROSIONE CHIMICA è una reazione eterogenea di interazione tra la fase liquida (l) o gassosa (g) con la fase solida (s) - metallo e si divide in 2 tipologie (corrosione gassosa, corrosione in liquidi non elettrolitici). 2.1. Corrosione da gas In pratica è la CORROSIONE DA GAS ad avere la massima importanza. Molto spesso si manifesta come corrosione di materiali metallici ad alte temperature (decine di volte superiori al punto di ebollizione dell'acqua) in un'atmosfera di gas contenenti ossigeno. Per questo motivo si chiama CORROSIONE AD ALTA TEMPERATURA o OSSIDAZIONE AD ALTA TEMPERATURA. Sulla superficie dei prodotti realizzati con metalli e leghe, a seguito della loro interazione con un ambiente così corrosivo, si forma una pellicola di prodotti che rappresenta vari ossidi metallici. Allo stesso tempo, a seconda delle condizioni operative, si possono formare altri composti, ad esempio metalli solforati5. Ad esempio, nell'industria della raffinazione del petrolio, la corrosione del gas colpisce, ad esempio, i tubi dei forni, le pareti dei reattori, le colonne di reazione e altre apparecchiature esposte a idrogeno solforato, monossido di carbonio, azoto, ossigeno e altri gas ad alte temperature. Il termine "corrosione da gas" si riferisce alla distruzione dei metalli causata dall'azione di vapori e gas ad alte temperature, sebbene possa essere applicato anche a temperature più basse a 5 ZOLFO METALLI, gli stessi dei solfuri. 12 garantire che una pellicola di liquido che conduce corrente elettrica non si condensi sul metallo. Sulla superficie dei prodotti realizzati con metalli e leghe, a seguito della loro interazione con un ambiente corrosivo, si forma una pellicola di prodotti che rappresentano vari composti metallici. In generale, il processo di corrosione chimica può essere descritto, ad esempio, dalla reazione di ossidazione del metallo con gas riscaldato (T >>373K) (G2): 2Ме + nГ2 → 2Men+Гn. In un ambiente ricco di ossigeno, tale processo può essere descritto dalla seguente equazione Me(T) + ½ O2 ↔ MeO(T). Questa reazione è equilibrio alle alte temperature. Il processo inverso è chiamato DISSOCIAZIONE DELL'OSSIDO. L'ossigeno presente in un ambiente gassoso è caratterizzato dalla sua pressione parziale (pO2). La reazione chimica di ossidazione del metallo sarà in equilibrio con la reazione di dissociazione dell'ossido se la pressione parziale dell'ossigeno (pO2) e l'ELASTICITÀ DI DISSOCIAZIONE DELL'OSSIDO (pMeO)7 diventano uguali (pMeO = pO2) 1. Quando pO2 > pMeO, la reazione procede principalmente nella direzione della formazione dell'ossido. Quando pMeO > pO2 la reazione procede prevalentemente nella direzione opposta. L'ossidazione dei metalli è un processo eterogeneo a più fasi, grazie al quale sulla superficie si forma prima uno strato di ossido mono e poi uno strato polimolecolare, il cosiddetto film di ossido. La crescita del suo spessore può avvenire sia in entrambe le direzioni contemporaneamente (mezzo metallico e gassoso), sia prevalentemente in una (Fig. 3). PRESSIONE PARZIALE (dal tardo latino partialis - parziale), la pressione che avrebbe un gas compreso in una miscela di gas se occupasse da solo un volume pari al volume della miscela alla stessa temperatura. 7 La pressione parziale di equilibrio dell'ossigeno per le reazioni di formazione o DISSOCIAZIONE TERMICA degli OSSIDI è detta ELASTICITÀ DI DISSOCIAZIONE DEGLI OSSIDI. Il suo valore è legato alla costante di equilibrio della reazione e, quindi, è funzione della temperatura. 6 13 abcFig. Fig. 3. Schema del meccanismo ionico-elettronico di formazione e crescita di un film di ossido sulla superficie di una parte metallica Se la crescita avviene nella direzione di un mezzo gassoso, si osserva un aumento significativo delle dimensioni della parte , ad esempio, durante l'ossidazione, di cui si deve tenere conto nell'ingegneria meccanica. La direzione di crescita è determinata dal rapporto tra i tassi di controdiffusione degli ioni metallici Мen+ e dell'ossigeno O2− all'interno della pellicola di ossido. Se le velocità di diffusione degli ioni metallici (rdif.Me) e dell'ossigeno (rdif.O2) differiscono notevolmente, la crescita della pellicola di ossido avviene prevalentemente in una direzione, ad esempio con rdiff.O2< rдиф.Mе − в направлении газовой среды и наоборот (рис. 3, в, б). В большинстве случаев, скорости диффузии ионов металла и кислорода соизмеримы, но для ионов кислорода скорость несколько ниже из−за того, что они больше по размерам, поэтому зона роста, находящаяся в толще самой оксидной пленки, располагается ближе к ее внешней поверхности (рис. 3, а). По мере утолщения оксидной пленки процессы встречной диффузии ионов и электронов затрудняются. С повышением температуры упругость диссоциации оксидов возрастает у всех металлов. Поэтому, начиная от какой−то температуры, когда упругость диссоциации оксида металла становится больше парциального давления кислорода в воздухе (при атмосферном давлении рО2 = 0,21 ат), металл перестает окисляться и становится вполне малоактивным по отношению к кислороду. Так, например, серебро при температуре 300 ºК термодинамически еще не устойчиво, а при 400 ºК и при всех более высоких температурах упругость диссоциации Ag2O превышает парциаль14 ное давление кислорода в воздухе. При температуре 2000 ºК медь тоже становится неокисляемым металлом, однако для таких металлов, как Fе, Zn, Ni и др., даже при этих температурах упругость диссоциации окислов остается еще достаточно низкой и, следовательно, протекание реакции окисления вероятно. Необходимо учесть, что температурная зависимость для кинетики процесса окисления совершенно иная, чем для термодинамической вероятности окисления. Поэтому нет никакого противоречия в том, что термодинамическая вероятность окисления металла с повышением температуры падает, а реальная скорость окисления (при условии, что термодинамически этот процесс остается еще возможным) с повышением температуры возрастает. 2.2. Образование тонких пленок на металлах Ранее было отмечено, что в большинстве случаев образовавшиеся продукты газовой коррозии – оксиды металла остаются на поверхности металла в виде пленки. Эти пленки определяют кинетику процесса и в случае наличия защитных свойств могут привести к замедлению коррозии. Чтобы пленка оксида металла обладала защитными свойствами, она должна быть: − сплошной; − прочно сцепляться с основным металлом и обладать близким к нему коэффициентом теплового расширения; − быть химически инертной по отношению к данной агрессивной среде; − обладать твердостью и износостойкостью. Даже при обычной температуре на поверхности многих металлов и сплавов на воздухе образуются слои оксидов. Еще в 1836 г. М. Фарадей высказал предположение, что даже на блестящей поверхности металлов существует тончайшая, невидимая защитная (оксидная) пленка. Этим он объяснял пассивность железа в азотной кислоте. Если пленка пористая, рыхлая и характеризуется плохим сцеплением с основным металлом, то даже при условии инертности в данной агрессивной среде, она не будет обладать защитными свойствами. Пленки по толщине (δ) делятся на три группы: − тонкие (невидимые), δ < 4000 нм; − средние (дающие цвета побежалости), δ = от 4000 до 50000 нм; 15 − толстые (видимые), δ > 50000nm. Lo spessore delle pellicole formate durante l'interazione del metallo con l'aria secca è diverso e dipende dal tipo di metallo, dalla temperatura e da altri fattori. Pertanto, lo spessore delle pellicole su rame e ferro a temperatura ambiente è 100–300 nm e su alluminio 1000–1500 nm. Diamo alcuni esempi che dimostrano la presenza di film sottili sui metalli. 1. Quando la superficie pulita e lucida di una piastra di ferro viene riscaldata a un'estremità, su di essa appariranno dei colori di interferenza: i "colori ossidanti". Ciò indica la comparsa di ossidi di vario spessore. I colori vanno nel seguente ordine: dall'estremità fredda del piatto a quella calda, rispettivamente: giallo, arancione, rosso, viola, viola, blu. Quando si testano le proprietà protettive del film formato utilizzando gocce di una soluzione di nitrato di rame, nei punti in cui il film ha proprietà protettive deboli, reagisce con il ferro: Cu(NO3)2 + Fe → Fe(NO3)2 + Cu. Di conseguenza, il rame apparirà sulla superficie sotto le gocce. Dopo il tempo in cui avviene la reazione, si possono giudicare le proprietà protettive delle pellicole in diverse aree. In questo esperimento, le migliori proprietà protettive si riscontrano nell'area che si trova lontano dal punto di riscaldamento (dietro la zona gialla), dove la superficie rimane invariata. È ovvio che in questa zona è presente una pellicola molto sottile, ma più densa rispetto alle zone riscaldate. 2. Se si rompe un ago (o una lama di rasoio) sotto il mercurio, nei punti rotti si formerà dell'amalgama. Quando i campioni frantumati vengono posti in aria nel mercurio, non si osserva alcuna amalgama. Ciò si spiega con la formazione di una pellicola di ossido molto sottile, che impedisce l'amalgamazione. 3. Quando il ferro viene posto in una soluzione di idrossido di potassio con eccesso di iodio, sulla superficie del ferro nella soluzione viene praticato un graffio profondo. Dopo un po ', il ferro nel punto del graffio inizia a dissolversi e sul fondo del vetro rimangono delle scaglie, che dopo un ulteriore esame risultano essere ossido di ferro (III). Il processo di formazione del film sul metallo avviene in più fasi. 16 La prima fase del processo di interazione di un metallo con un ambiente corrosivo è il chemiassorbimento dell'agente ossidante (O2, CO2, H2O, SO2) sulla superficie del metallo, ad esempio: Me(sol) + (O2) = Me( sol) + 2Oads. In presenza di affinità chimica tra il metallo e l'agente ossidante, si realizza il secondo stadio di interazione del metallo con l'ambiente corrosivo: la transizione del film chemisorbito a quello di ossido. Questo processo può essere convenzionalmente descritto da una reazione del tipo: xMe(s) + yOads = MexOy. La pellicola di ossido formata sulla superficie del metallo può rallentare il processo di corrosione a causa dell'inibizione dell'apporto dell'agente ossidante alla superficie del metallo ossidante. In questo caso la pellicola HA PROPRIETÀ PROTETTIVE. 2.2.1. Condizione per la continuità dei film sui metalli Solo i FILM CONTINUI possono avere proprietà protettive. La possibilità della formazione di tale pellicola è determinata dalla CONDIZIONE DI CONTINUITÀ di Pilling-Bedworth. Il volume molare del composto che appare sulla superficie del metallo (Vok) deve essere maggiore del volume del metallo (VMe) consumato per formare una mole del composto: Vok/VMe >1. Se Vok/VMe< 1, то пленка не может быть сплошной. К металлам, не удовлетворяющим условию сплошности при окислении их кислородом, относятся щелочные и щелочно−земельные металлы. Сплошность пленок – необходимый, но недостаточный фактор, определяющий ее защитные свойства. Как показал Францевич И. Н., у пленок с Vok/VMe >>1 non può avere proprietà protettive elevate, a causa della presenza di grandi tensioni interne in essi (ad esempio MoO3 o WO3). Il limite superiore del rapporto volumetrico è considerato pari a 2,5. Allora la condizione di continuità raffinata è la seguente: 1< Vok/VMe < 2,5. 2.2.2. Законы роста оксидных пленок на металлах Процесс роста ПОРИСТОЙ ПЛЕНКИ не осложняется процессом подвода окислителя к поверхности металла, а контролируется скоростью химической реакции образования оксида. Скорость коррозии в этом случае выражается уравнением: 17 V = dx/d = kC, где x − толщина пленки; – время; k − константа скорости химической реакции образования оксида; C − концентрация окислителя на поверхности метала. Рост пористой пленки, контролируемый скоростью химической реакции окисления металла, протекает во времени ПО ЛИНЕЙНОМУ ЗАКОНУ. Линейный закон роста имеет место: − при высокотемпературном окислении на воздухе и в среде кислорода при Vok/VMe<1; − в случае образовании летучих оксидов. Сплошные пленки затрудняют проникновение реагентов друг к другу и их рост сопровождается самоторможением процесса. Результирующая скорость этого сложного процесса определяется скоростью самой медленной стадии, т.е. возможны различные варианты контроля течения процесса. 1−й вариант. Скорость коррозии контролируется стадией массопереноса (диффузионный контроль процесса). В этом случае концентрация окислителя на границе раздела Ме – оксидная пленка равна нулю, так как скорость химической реакции весьма велика. Уравнение скорости течения коррозии определяется ПАРАБОЛИЧЕСКИМ ЗАКОНОМ роста оксидной пленки. Параболический закон роста реализуется, в частности, при окислении железа на воздухе при различных температурах. 2−ой вариант. В предыдущем случае С = 0, т.е. накопление окислителя на внутренней поверхности оксидной пленки не происходит. Если учесть возможность кинетического торможения процесса роста оксидной пленки наряду с торможением на стадии массопереноса (диффузионно−кинетический контроль процесса), в этом случае приходим к СЛОЖНО − ПАРАБОЛИЧЕСКОМУ ЗАКОНУ роста оксидной пленки. Рост оксидных пленок по сложно−параболическому закону выполняется: а) при окислении железа в парах воды или в атмосфере СО2; б) при окислении Сo, Сu в атмосфере чистого кислорода; в) при частичном разрушении оксидной пленки. Рост оксидных пленок при диффузионно−кинетическом контроле может быть выражен ТАКЖЕ СТЕПЕННЫМ ЗАКОНОМ. 18 3−й вариант. Часто рост пленки протекает медленнее, чем это следует из параболического закона роста. Это наблюдается при возникновении оксидных пленок при низких температурах (на Сu в О2 при t < 100 оС; на Аl, Тi, Ni, Zn в О2 при t = 300400 оС.) Затухание объясняется либо уплотнением пленок, либо появлением дефектов (пузырей, расслоений). В этих случаях рост толщины пленки протекает в соответствии с логарифмическим законом роста: x = ln(K). 2.3. Термодинамика процесса химической коррозии В качестве примера рассмотрим процесс окисления металла в атмосфере кислорода. Процесс окисления металла может быть представлен реакцией вида: 2Ме + nО2 → Me2n+Оn. Движущая сила процесса коррозии (ДСП) может быть определена с помощью термодинамики. Торможение процесса (ТП) не может быть определено термодинамически, однако с помощью термодинамики можно оценить условия, уменьшающие или исключающие протекание процесса (применение защитных сред, катодная защита, обескислороживание и др.). Независимо от механизма коррозии возможность ее протекания определяется знаком изменения термодинамического потенциала (см. раздел 1.1). Любое изменение энергии системы характеризует переход ее в новое состояние и является мерой стабильности системы: ΔG = GII − GI , где ΔG – энергия, израсходованная на изменение состояния системы, например, на процесс коррозии; GI − энергия системы в исходном состоянии, например, металла в конкретных условиях; G II − энергия системы в новом состоянии, например, прокорродировавшего металла. Рассчитать ΔG можно, вспомнив начало химии8. Величину изобарно-изотермического потенциала можно рассчитать по разности между суммами Δ G продуктов реакции и исходных веществ: GХР = ΔG ni – ΔG mi. (прод. р-ции) (исх. в-в). ΔG - является функцией температуры. Стандартные значения для большинства веществ сведены в справочные таблицы термодинамических величин. Изменение энергии Гиббса в зависимости от температуры учитывается уравнением Гиббса. ΔG Т = ΔН 298 – 298 Δ S 298. ΔG простого вещества = 0. Определить энергию Гиббса для любой химической реакции можно по уравнению ΔG = ΔG + RТ ln К, рассчитав предварительно величину константы равновесия, например, по равновесным концентрациям веществ. При равновесии ΔG=0, поэтому ΔG+RТlnК=0, следова8 19 2.4. Кинетика процесса химической коррозии Хотя термодинамика и дает ответ на вопрос о том, насколько изучаемая система отдалена от состояния равновесия, но ответ на весьма важный практический вопрос о скорости протекания коррозионного разрушения в рамках термодинамики получен быть не может. Рассмотрением этого вопроса и занимается кинетика коррозионных процессов. 2.4.1. Скорость процесса коррозии. Показатели химической коррозии СКОРОСТЬ КОРРОЗИОННОГО процесса может быть представлена в общем виде с помощью уравнения: СКОРОСТЬ КОРРОЗИИ (СК) = ДСП / ТП, где ДСП − движущая сила процесса; ТП − торможение процесса. Скорость химической коррозии определяют количественно по наблюдениям во времени за изменением какой−либо подходящей для этих целей величины, изменяющейся в процессе коррозии. Тогда истинная скорость коррозии металлического материала определяется из выражения: V = dy/d, где y − изменяющаяся в процессе коррозии характеристика свойств материала; − время коррозии. Для количественного выражения скорости коррозионных разрушений на практике пользуются показателями коррозии, являющимися, по сути, отражением СРЕДНЕЙ СКОРОСТИ КОРРОЗИОННОГО разрушения материала: Vср=∆y/∆. Показатели коррозии. 1. ГЛУБИННЫЙ ПОКАЗАТЕЛЬ коррозии: Kn=∆П/∆, где ∆П − глубина (средняя или максимальная) коррозионного разрушения; ∆ − промежуток времени коррозии. 2. ПОКАЗАТЕЛЬ ИЗМЕНЕНИЯ ТОЛЩИНЫ образующейся на металле пленки продуктов коррозии: Kn= ∆h/∆, где ∆h − изменение толщины образующейся на металле пленки продуктов коррозии. тельно, ΔG = – RТlnК. Данное уравнение позволяет также определить значение константы равновесия химической реакции по величине изобарно-изотермического потенциала. Для определения направления протекания многих химических реакций достаточно оценить знак Δ G. 1). При ΔН 0 , Δ S 0 реакция самопроизвольна (Δ G 0) при Т. 2). При ΔН 0, Δ S 0 реакция обратная (Δ G 0) при любой Т. 3). При ΔН 0, Δ S 0, реакция самопроизвольна (Δ G 0) при любой Т. 4). При ΔН 0, ΔS 0 реакция самопроизвольна (Δ G 0) при Т. 20 3. ПОКАЗАТЕЛЬ ИЗМЕНЕНИЯ МАССЫ Km=∆m/S∆, где ∆m − изменение массы корродирующего металла; S – площадь поверхности коррозии. 4.ОБЪЕМНЫЙ ПОКАЗАТЕЛЬ КОРРОЗИИ Kv=∆V/S∆, где ∆V − объем газа, выделившегося или поглощенного в процессе коррозии и приведенный к нормальным условиям. 5. МЕХАНИЧЕСКИЙ ПОКАЗАТЕЛЬ КОРРОЗИИ Кs=(∆s/∆)100 %, где ∆s − относительное изменение характеристики механического свойства. 6. ПОКАЗАТЕЛЬ ИЗМЕНЕНИЯ ЭЛЕКТРИЧЕСКОГО СОПРОТИВЛЕНИЯ KR=(∆R/∆)100 %, где ∆R − относительное изменение электросопротивления образца. 2.4.2. Влияние внешних факторов на химическую коррозию металлов Скорость и характер процесса химической коррозии металлов зависит от ряда факторов. ВНЕШНИМИ ФАКТОРАМИ называют те, которые связаны с составом коррозионной среды и условиями коррозии (температура, давление, скорость перемещения коррозионной среды и т.д.). Температура − мощный внешний фактор коррозии. Характер влияния температуры на скорость окисления металла определяется зависимостью константы скорости реакции окисления (k) и диффузии (D) от температуры. И k = f(T) и D = f(T) описываются одним и тем же уравнением (уравнение Аррениуса): k = k0 exp (−E/RT), (9) где k0 − константа; Е − энергия активации химической реакции. D = D0 exp (−E1/RT), (10) 1 где D0 – константа; Е − энергия активации диффузии. Таким образом, вне зависимости от вида контролирующей стадии процесса окисления с повышением температуры скорость окисления резко возрастает. Колебания температуры, особенно переменный нагрев и охлаждение, увеличивают скорость окисления металла, так как в защитной пленке образуются трещины. Состав газовой фазы. Влияние состава газовой фазы на скорость коррозии металла велико, специфично и изменяется с изменением температуры. В частности, на скорость окисления железа и 21 стали особенно сильно влияют кислород, соединения серы, водяные пары. Приведенные ниже данные свидетельствуют о зависимости увеличения относительной скорости коррозии стали с 0,17 % углерода от состава газовой фазы при 900 оС: Состав газовой фазы Кислород Чистый воздух Чистый воздух + 2 % SO2 Чистый воздух + 5 % H2O Чистый воздух + 5 % SO2+ 5 % H2O % 200 100 118 134 276 Значительное влияние на коррозию сталей и сплавов оказывают продукты горения топлива, содержащие ванадий (например, V2O5). Это соединение находится в золе от сжигания дешевого топлива − мазута, нефтепродуктов. Зола, налипая на металл, увеличивает скорость его коррозии в десятки раз. Причина этому – «ванадиевая коррозия», обусловленная легкоплавкостью V2O5, и его способностью ОФЛЮСОВЫВАТЬ (переводить в жидкое состояние) химические соединения золы и окалины. Пятиокись ванадия участвует в процессе окисления железа: 4Fe + 3V2O5 = 2Fe2O3 + 3V2O3, V2O3 + O2 = V2O5, V2O5 + Fe2O3 = 2FeVO4. Повышение содержания СО в газовой фазе понижет скорость коррозии углеродистых и низколегированных сталей, но при больших количествах СО в газовой фазе может произойти науглероживание поверхности стали. При этом возможны следующие химические реакции: 2CO + O2 = 2CO2, 2CO = Cсаж + CO2. Давление окислителя. Если бы поверхность металла не была покрыта оксидной пленкой, то скорость окисления была бы пропорциональна парциальному давлению окислителя (для двухатомных газов – PГ2). Так как поверхность металла покрыта слоем оксида, то зависимость скорости окисления от величины парциального давления окисляющего газа определяется строением защитной пленки. В то же время при окислении железа при 700 − 950о С скорость окисления не зависит от парциального давления Po2. 22 Скорость движения газовой фазы. Окисление как гетерогенный процесс определяется скоростью подвода и отвода реагентов в зону реакции. Поэтому, чем больше скорость движения потока газа, тем больше и скорость окисления металла. Режим нагрева металла. Влияние режима нагрева металла может быть рассмотрено в зависимости от влияния колебаний температуры. Иначе говоря, переменные нагрев и охлаждение увеличивают скорость окисления ввиду нарушения сохранности защитной пленки. 2.4.3. Влияние внутренних факторов на химическую коррозию металлов ВНУТРЕННИМИ называют факторы, связанные с составом, структурой, внутренними напряжениями в металле, качеством обработки поверхности металла и др. Состав сплава. Применительно к наиболее важным конструкционным сплавам на основе железа для случая газовой коррозии можно отметить следующее. 1. При высоких температурах (более 800оС) с увеличением содержания углерода в стали скорость ее окисления и обезуглероживания уменьшается вследствие более активного образования СО, что снижает окислительный потенциал газовой фазы. 2. Сера фосфор, никель, марганец практически не влияют на скорость окисления железа. 3.Титан, медь, кобальт, бериллий заметно снижают скорость окисления железа. 4. Хром, алюминий, кремний сильно замедляют окисление железа. 5. Ванадий, вольфрам и молибден могут значительно ускорить окисление стали, которое иногда носит катастрофический характер. Структура сплава. Анализ экспериментальных данных свидетельствует о том, что чем меньше в сплаве структурных составляющих, тем выше его жаростойкость. Применительно к сплавам железо – углерод наиболее устойчивой является АУСТЕНИТНАЯ СТРУКТУРА9. Аустенит (γ-фаза) − высокотемпературная гранецентрированная модификация железа и его сплавов. В углеродистых сталях аустенит − это твѐрдый раствор внедрения, в котором атомы углерода входят внутрь элементарной ячейки γ-железа во время конечной термообработки. В сталях, содержащих другие металлы (кроме железа, легированные стали), атомы металлов замещают атомы железа в кристаллической решетке, и возникает твердый раствор замещения. Фаза названа в честь сэра Уильяма Чандлера Робертс-Остина (англ. William Chandler Roberts-Austen, 1843 − 1902). 9 23 Всего различают пять больших групп нержавеющих сталей, определяемых их микроструктурой. Наиболее распространенными являются три: АУСТЕТНИТНЫЕ (AUSTENITIC) − не магнитная сталь с основными составляющими 15−20 % хрома и 5−15 % никеля, который увеличивает сопротивление коррозии. Она хорошо подвергается тепловой обработке и сварке. Обозначается буквой A в начале наименования марки. Именно аустенитная группа сталей наиболее широко используется в промышленности и в производстве элементов крепежа. МАРТЕНСИТНЫЕ (MARTENSITIC) − значительно более твердые, чем аустетнитные стали и могут быть магнитными. Они упрочняются, закалкой и отпуском подобно простым углеродистым сталям и находят применение главным образом в изготовлении столовых приборов, режущих инструментов и общем машиностроении. Больше подвержены коррозии. Обозначаются буквой С. ФЕРРИТНЫЕ (FERRITIC) стали значительно более мягкие чем мартенситные по причине малого содержания углерода. Они также обладают магнитными свойствами. Обозначаются буквой F. Стали с двухфазной АУСТЕНИТНО−ФЕРРИТНОЙ СТРУКТУРОЙ менее устойчивы против окисления. Их меньшая жаростойкость связывается с большей неоднородностью образующейся защитной пленки, что приводит к ее разрушению при росте (неоднородность возникающих внутренних напряжений). Так хромоникелевые стали с однофазной аустенитной структурой более устойчивы против окисления, чем двухфазные: Х12Н12М2Т, Х12Н9Т ведут себя лучше, чем ОХ21Н5МД2Т или 1Х21Н5Т 2]. На жаростойкость10 чугунов кроме их состава влияет и структура. Существенное влияние оказывает форма графитных включений. Так, при шаровидной форме графита стойкость против окисления выше, чем при пластинчатой. Установлено, что предварительная холодная пластичная деформация несколько ускоряет окисление металла вследствие повышения запаса его энергии. Чем тщательнее обработана поверхность металла, тем меньше скорость его окислеЖаростойкость − окалиностойкость, способность металлических материалов противостоять химическому разрушению поверхности под воздействием воздушной или иных газообразных сред при высоких температурах. 10 24 ния, что обусловлено лучшей сохранностью защитных пленок на гладкой поверхности. 2.5. Некоторые случаи газовой коррозии металлов в технологических средах В химической промышленности многие технологические процессы или их определенные стадии протекают в газовой среде в условиях повышенных температур и давлений (рис.4). При температурах от 100 до 200 − 300 °С многие газы не опасны. Химическая активность газов и скорость газовой коррозии металлов сильно возрастают при температурах выше 200 − 300 °С. Так, Cl2 начинает действовать на железные сплавы при Рис. 4. Некоторые случаи газовой температуре > 200 °C, HCl -> corrosione dei metalli in ambito tecnologico - 300 °C, SO2, NO2, vapori di zolfo. > 500 °C. Queste caratteristiche del comportamento degli ambienti gassosi tecnologici e il loro uso diffuso nell'industria richiedono una considerazione più dettagliata del comportamento dei metalli in condizioni reali. 2.5.1. Corrosione di ferro, ghisa e acciai in un'atmosfera di vapori di O2, CO2, H2O L'impatto dell'ossigeno dell'aria ad alte temperature sulle leghe ferro-carbonio porta all'ossidazione del ferro con formazione di incrostazioni, decarburazione dell'acciaio e crescita. di ghisa. A seguito dell'ossidazione del ferro ad alte temperature si forma uno strato di prodotti di corrosione chiamato SCALE. L'incrostazione ha una struttura complessa e comprende diversi ossidi: Fe3O4 – MAGNETITE ha un reticolo cristallino di spinello complesso; Fe2O3 – L’EMATITE ha un reticolo romboedrico; FeO – VUSTIT ha una struttura reticolare cristallina difettosa. La wustite si forma a temperature superiori a 575 oC e si decompone con raffreddamento lento: 4FeO Fe3O4 + Fe. 25 Al di sotto di 575 °C non è presente wustite nell'incrostazione e uno strato di Fe3O4 è direttamente adiacente alla superficie dell'acciaio. La velocità di ossidazione dell'acciaio (vedi Sezione 2.4.1) aumenta con l'aumentare della temperatura secondo una legge prossima all'esponenziale (Fig. 5), e nelle coordinate logk − 1/T è espressa da una linea spezzata, ciascuna interruzione della quale corrisponde a qualche trasformazione. Pertanto, a una temperatura di 575 ºC, nella scala appare un sottostrato di wustite FeO, che non interferisce con la diffusione dell'ossigeno, a seguito del quale aumenta l'energia di attivazione del processo e aumenta la velocità di ossidazione del metallo. L'interruzione della curva ad una temperatura di ~ 900 ºС corrisponde alla trasformazione allotropica dell'acciaio. La natura del cambiamento nella dipendenza dalla temperatura del tasso di ossidazione dell'acciaio indica che la struttura austenitica è più resistente al calore, in cui si osserva un aumento più lento del tasso di ossidazione all'aumentare della temperatura. Insieme all'ossidazione negli acciai e nella ghisa, si verifica il processo di decarbonizzazione: l'esaurimento dello strato superficiale di carbonio a causa dell'interazione del carburo di ferro in essi contenuto con l'ossigeno e i reagenti contenenti ossigeno Fe3C+O2→3Fe+CO2 Fe3C+CO2 →3Fe+2CO Fe3C+H2O→3Fe+CO + H2. 26 La composizione dell'ambiente gassoso ha una forte influenza sul tasso di ossidazione dell'acciaio e della ghisa. Nella corrosione dei gas, gli agenti aggressivi possono essere, ad esempio, cloro, composti di zolfo, ossigeno, aria, composti di iodio, ecc. La loro aggressività nei confronti dei diversi metalli non è la stessa, quindi la velocità di corrosione varia. Pertanto, il tasso di ossidazione di Fe, Co, Ni alla temperatura di 900 °C aumenta nell'ordine H2O → CO2 → O2 → SO2. In questo caso i metalli, a seconda della velocità di corrosione nell'atmosfera di questi reagenti, sono disposti in serie ascendente: Ni → Co → Fe. Se assumiamo che il tasso di corrosione nell'aria a 900 °C sia del 100%, l'aggiunta di 2% SO2 aumenta questo tasso al 118%, 5% H2O al 134%, 2% SO2 + 5% H2O al 276%. 2.5.2. Corrosione sotto l'influenza dei prodotti della combustione del carburante I prodotti della combustione del carburante (carbone, olio combustibile, ecc.) nella maggior parte dei casi contengono quantità significative di composti di zolfo (SO2, H2S) e vanadio sotto forma di V2O5, che è un forte agente ossidante. L'ossido di vanadio bassofondente interagisce con lo strato protettivo di incrostazioni sulla superficie del metallo, distruggendolo e formando vanadati, che creano eutettici11 con basso punto di fusione Fe2O3 + V2O5 → 2FeVO4 con ossido di vanadio V2O5. Pertanto, il vanadato di ferro (FeVO4) ha punto di fusione = 840 °C, e il punto di fusione del suo eutettico con V2O5 è 625 °C; Il punto di fusione del cromo vanadato CrVO4 è 810 °C e la sua eutettica con V2O5 è 665 °C. Quindi il vanadate partecipa attivamente al processo di ossidazione del metallo stesso 6FeVO4+4Fe→ 5Fe2O3 + 3V2O3 4Fe+ 3V2O5→ 2Fe2O3 + 3V2O3 V2O3+O2→ V2O5, e il V2O5 stesso non viene praticamente consumato. La corrosione da vanadio colpisce soprattutto le bobine, i supporti e le pareti divisorie dei forni che operano nell'intervallo di temperature compreso tra 620 e 700 °C. Eutettico (dal greco éutektos - facilmente fondente), sistema liquido (soluzione o fuso) che ad una data pressione è in equilibrio con fasi solide, il cui numero è pari al numero dei componenti del sistema. 11 27 Allo stesso tempo, il tasso di corrosione anche degli acciai al cromo e al cromo-nichel, che non sono soggetti alla corrosione da idrogeno solforato ad alta temperatura, raggiunge i 15 mm/anno. Sotto l'influenza dei composti dello zolfo, gli acciai ferro-carbonio sono soggetti ad un'intensa corrosione intergranulare a causa di un numero maggiore di difetti nei reticoli cristallini dei solfuri rispetto agli ossidi. Ciò porta all’intensificazione dei processi di diffusione. Con un aumento del contenuto di monossido di carbonio (II) nei prodotti della combustione, la velocità di corrosione del gas degli acciai al carbonio e a bassa lega diminuisce notevolmente. Tuttavia, la sua concentrazione molto elevata porta alla carburazione della superficie: 3Fe+2CO→Fe3С+CO2. Le leghe altamente legate del tipo X40N50 sono resistenti alla corrosione del vanadio. Anche l'introduzione del silicio nell'acciaio ha un effetto positivo; la lega dell'acciaio con molibdeno, tungsteno e vanadio ha un effetto negativo sulla durabilità dell'acciaio, poiché questi elementi di lega favoriscono12 la formazione di V2O5. Le leghe a base di rame sono altamente suscettibili a tale corrosione. Per ridurre la velocità di corrosione del vanadio attualmente si adotta: − limitare la temperatura di esercizio a 650 °C; − utilizzo di leghe del tipo X40N50 per la fabbricazione di componenti soggetti a questo tipo di distruzione; − insufflando nel combustibile polvere di dolomite contenente ossidi di magnesio e di alluminio, che formano composti refrattari con l'ossido di vanadio; − limitare il contenuto totale di sodio e vanadio nel carburante a non più del 210–4%. 2.5.3. Corrosione nell'ambiente di cloro e acido cloridrico Il comportamento dei metalli nell'ambiente di cloro gassoso e acido cloridrico è fondamentalmente diverso dall'azione di altri ambienti aggressivi. Ciò è dovuto al fatto che i sali cloruro che si formano sulla superficie del metallo hanno un basso punto di fusione e in alcuni casi, quando la temperatura aumenta, sublimano13, ad esempio Ti + 2Cl2 → TiCl4. Promuovere - (sinonimi) guidare, promuovere, dirigere. La sublimazione è il passaggio di una sostanza dallo stato solido a quello gassoso, bypassando la fase liquida; lo stesso della sublimazione. 12 13 28 La maggior parte di queste reazioni sono esotermiche. La velocità di rimozione del calore risulta essere inferiore alla velocità della reazione stessa, a seguito della quale i metalli si accendono e “bruciano” in un'atmosfera di cloro. Ciò porta ad un significativo aumento locale della temperatura e i risultanti cloruri si sciolgono e si decompongono. I più resistenti al cloro sono gli acciai al nichel, al piombo e al cromo. Temperatura di accensione di alcuni metalli in un'atmosfera di cloro secco: o t accensione. Con Ti< 20 Pb 90−100 Fe 150 Cu 200 Ni > 500. Nell'acido cloridrico secco a temperatura ambiente, numerosi metalli e leghe presentano una resistenza soddisfacente. Con l'aumentare della temperatura, la resistenza dei materiali metallici diminuisce gradualmente fino ad una temperatura specifica per ciascun metallo. Le temperature massime elevate consentite durante il funzionamento a lungo termine di metalli e leghe in cloro secco e acido cloridrico sono riportate nella tabella. 2. I metalli più resistenti nel cloro secco, ad eccezione dei metalli nobili, sono il nichel e le sue leghe. Le pellicole superficiali formate sugli acciai al nichel e al cromo-nichel hanno una bassa volatilità e proprietà protettive soddisfacenti. Tabella 2 Temperature consentite durante l'utilizzo di alcuni metalli e leghe in un'atmosfera di acido cloridrico e cloro3] T, °C Materiale Cl2 1200 − − 550 − 100 150 300 − Platino Oro Tungsteno Nichel Inconel (80% Ni, 14% Cr, 6% Fe) Rame Acciaio al carbonio St.Z Acciaio inossidabile 12Х18Н9Т Argento 29 HCl 1200 870 600 510−600 480 100−120 260−350 450−500 230 2.5.4. Corrosione da idrogeno dell'acciaio La corrosione da idrogeno può accompagnare molti processi tecnologici che si verificano a temperature elevate da 200°C e pressioni da 300 MPa in ambienti contenenti idrogeno. Queste condizioni corrispondono a processi come l'idrogenazione del carbone e del petrolio, la sintesi di ammoniaca e metanolo, ecc. Si osservano due tipi di danni ai metalli causati dall'idrogeno: FRAGILITÀ DELL'IDROGENO e CORROSIONE DELL'IDROGENO. Spesso questi fenomeni si sovrappongono. Se nel gas è presente ammoniaca si può verificare anche la NITRIDAZIONE DEL METALLO. Quando una miscela di azoto-idrogeno entra in contatto con un metallo in condizioni di temperatura e pressione elevate, l'idrogeno molecolare si dissocia sulla superficie del metallo. L'idrogeno atomico risultante si diffonde nel reticolo metallico e in esso si dissolve. Quando la temperatura diminuisce a causa della diminuzione della solubilità, l'idrogeno tende a passare allo stato gassoso all'interno del metallo. In questo caso, nel metallo si verificano grandi sollecitazioni che portano a fragilità irreversibile. La corrosione da idrogeno è il risultato dell'interazione chimica dell'idrogeno con il componente in carburo dell'acciaio. Esternamente, la manifestazione della corrosione da idrogeno significa una forte diminuzione della resistenza dell'acciaio senza una notevole distruzione della superficie. Il meccanismo della corrosione dell'idrogeno prevede le seguenti fasi: - ad alte temperature, l'idrogeno molecolare si dissocia in atomi che vengono assorbiti dalla superficie dell'acciaio e diffondono nel suo reticolo cristallino; − essendo un agente riducente attivo, l'idrogeno atomico riduce il carburo di ferro e interagisce con il carbonio disciolto nell'acciaio attraverso reazioni irreversibili: Fe3C + 2H2 3Fe + CH4 C + 4H CH4, privando così l'acciaio della sua base rinforzante. Poiché i processi di diffusione, compreso il movimento dell'idrogeno, si realizzano più facilmente lungo i bordi dei grani, dove si trovano prevalentemente le piastre di cementite, la loro distruzione da parte dell'idrogeno porta ad un'interruzione della connessione tra cristalliti e, di conseguenza, ad una diminuzione della duttilità dell'acciaio . 30 Il metano risultante ha una dimensione molecolare grande rispetto ai parametri del reticolo cristallino della ferrite. Di conseguenza, non può diffondersi dalla massa del metallo e si accumula nelle sue microcavità e nei suoi difetti, causando un'elevata pressione intracavità e portando alla fessurazione. In questo caso si sviluppano delle crepe lungo i bordi dei grani. Negli acciai dolci con proprietà a bassa resistenza, gli accumuli di metano si verificano prevalentemente nello strato vicino alla superficie, formando rigonfiamenti chiaramente visibili sulla superficie dell'acciaio. Oltre a partecipare alla formazione del metano, l'idrogeno ha una buona solubilità nel metallo. La concentrazione di idrogeno disciolto nel metallo dipende dalla pressione parziale dell'idrogeno all'interfaccia metallo-gas e può essere determinata dalla formula: = Ks p1/2, dove è la quantità di idrogeno disciolto nell'acciaio, cm3/ 100 grammi; p – pressione parziale dell'idrogeno, a; Ks – solubilità dell'idrogeno nell'acciaio a p = 1 a. Poiché la dissoluzione dell'idrogeno da parte di un metallo è un processo endotermico, la solubilità dell'idrogeno aumenta con l'aumentare della temperatura. Questo fenomeno è chiamato PERMEABILITÀ ALL'IDROGENO (VH) 4]. La permeabilità all'idrogeno dell'acciaio dipende dal contenuto di carbonio e di elementi di lega in esso contenuti e diminuisce all'aumentare del contenuto di carbonio, come evidenziato dai dati riportati in tabella. 3. Tabella 3 Effetto del contenuto di carbonio sulla permeabilità all'idrogeno degli acciai al carbonio Metallo C, % in peso VН, cm3/cm2h mm–1 Ferro tecnico 0,04 5,10 Acciaio 15 0,15 4,09 Acciaio 30 0,27 2,79 Acciaio U10A 1,10 2,24 La corrosione da idrogeno non appare immediatamente dal momento in cui l'acciaio viene esposto all'idrogeno, ma dopo un certo periodo di tempo. Questo periodo, durante il quale non si verificano cambiamenti nella microstruttura e nelle proprietà meccaniche dell'acciaio, è chiamato PERIODO DI INCUBAZIONE DEL PROCESSO DI DECARBARATURA DELL'ACCIAIO. 31 Il periodo di incubazione è di grande importanza pratica, poiché determina essenzialmente il tempo di funzionamento sicuro dell'apparecchiatura. Il periodo di incubazione dipende principalmente dalla temperatura e dalla pressione. Con l'aumento della temperatura e della pressione, il periodo di incubazione diminuisce. Allo stesso tempo, la durata del periodo di incubazione è fortemente influenzata dalla composizione chimica dell'acciaio. La temperatura e la pressione parziale dell'idrogeno influenzano non solo la durata del periodo di incubazione, ma anche la velocità di decarburazione dell'acciaio durante la corrosione da idrogeno. 2.5.5. Corrosione carbonilica La corrosione carbonilica si verifica nei processi tecnologici che coinvolgono il carbonio a pressioni e temperature elevate. Tali processi includono, ad esempio, la produzione di alcoli metilico e butilico, la conversione di metano e monossido di carbonio. In condizioni normali, la CO è inerte nei confronti dei metalli. A temperature e pressioni elevate, il monossido di carbonio reagisce con molti metalli e forma carbonili. Ad esempio Fe + nCO = Fe(CO)n. Ferro e CO possono formare tre composti: tetracarbonile - Fe(CO)4, pentacarbonile - Fe(CO)5 e nanocarbonile - Fe(CO)g. Tutti questi composti sono piuttosto instabili e si decompongono quando la temperatura aumenta. Il composto più stabile tra questi è Fe(CO)5. La formazione di carbonili aumenta all'aumentare della temperatura a partire da 100 ºС, poi, nell'intervallo di temperature 140 – 180 ºС, si osserva la loro dissociazione quasi completa in Fe e CO. Il monossido di carbonio (II) può formare composti simili con molti altri metalli. La corrosione carbonilica provoca la distruzione e l'allentamento dello strato superficiale del metallo fino a una profondità di 5 mm. Non si verificano più cambiamenti nella struttura del metallo a una distanza maggiore dalla superficie. 2.5.6. Corrosione nei non elettroliti I NON ELETTROLITI sono liquidi che non conducono elettricità. Mezzi liquidi corrosivi di origine inorganica: bromo liquido, zolfo fuso, ecc. Sostanze organiche liquide: benzene, cloroformio, ecc., combustibile liquido (olio, cherosene, benzina, ecc.), oli lubrificanti. Nella loro forma pura sono poco aggressivi, tuttavia, la presenza in essi anche di piccole quantità di impurità (mercaptani14, idrogeno solforato, acqua, ossigeno, ecc.) Aumenta notevolmente la loro attività chimica. Pertanto, i mercaptani e l'idrogeno solforato contenuti nel petrolio greggio provocano la corrosione di Fe, Cu, Ni, Ag, Pb, Sn con la formazione rispettivamente dei loro mercaptidi (Me−(S−R)n) e polisolfuri (Me2Sx). Tracce di acqua nel tetracloruro di carbonio aumentano la sua corrosività a causa della formazione di componenti conduttivi (HCl) a seguito dell'idrolisi e del verificarsi di corrosione mediante meccanismo elettrochimico: CCl4 + H2O → COCl2 + 2HCl CCl4 + 2H2O + kаt → CO2 + 4HCl Principalmente la corrosione dei metalli nei non elettroliti procede attraverso un meccanismo chimico. Questo processo è composto da più fasi, ciascuna delle quali determina la velocità di corrosione: 1) diffusione dell'ossidante sulla superficie metallica; 2) chemiassorbimento dell'agente ossidante; 3) reazione chimica; 4) desorbimento dei prodotti della corrosione dalla superficie metallica; 5) diffusione dei prodotti della corrosione nel volume del non elettrolita. In alcuni casi, sulla superficie metallica si forma una pellicola protettiva dai prodotti della corrosione, che porta all'inibizione del processo di corrosione a causa della difficoltà di diffusione dell'ossidante sulla superficie metallica. A seconda delle proprietà protettive del film risultante e della sua solubilità, è possibile installare il controllo della diffusione, cinetico o misto-diffusione-cinetico del processo. La temperatura influenza in modo significativo il tasso di corrosione chimica dei metalli nei non elettroliti (vedere sezione 2.4.). L'effetto della temperatura sulla velocità del processo è in alcuni casi complicato dai cambiamenti nella solubilità dell'ossidante e della pellicola di prodotti della corrosione metallica nel non elettrolita quando la temperatura cambia. 14 I mercaptani sono derivati idrosolfuri degli idrocarburi: R – S –H. 33 La presenza di acqua nei non elettroliti attiva in modo significativo l'effetto corrosivo delle impurità e provoca un'intensa corrosione elettrochimica dei metalli, ad es. il meccanismo del processo di corrosione cambia. Per proteggere i metalli presenti nei non elettroliti dalla corrosione chimica, vengono selezionati metalli e leghe stabili in un dato ambiente (ad esempio, l'alluminio e le sue leghe sono stabili nella benzina crackizzata) e vengono applicati rivestimenti protettivi (ad esempio, l'acciaio in un ambiente l'ambiente di idrogeno solforato è rivestito di alluminio). 2.6. Metodi per proteggere i metalli da vari tipi di corrosione da gas 2.6.1. Metodi per proteggere l'acciaio dalla corrosione del gas Per proteggere l'acciaio dalla corrosione del gas, l'ALOYING resistente al calore e i RIVESTIMENTI PROTETTIVI sono i più utilizzati. Lega - (tedesco legieren - "fondere", dal latino ligare - "legare") - aggiungere impurità alla composizione dei materiali per modificare (migliorare) le proprietà fisiche e/o chimiche del materiale di base. I principali metodi per proteggere i metalli dall'ossidazione ad alte temperature si basano sull'ALLOYING. La resistenza dei metalli ossidati alle alte temperature dipende dalle proprietà protettive del film di ossido che ricopre il metallo. Gli elementi che contribuiscono alla creazione di uno strato protettivo sulle leghe ferro-carbonio sono il cromo, l'alluminio e il silicio. Questi elementi si ossidano più facilmente alle alte temperature nell'aria rispetto al metallo legato e formano scaglie più stabili. Secondo la teoria della lega resistente al calore, il componente legante forma sulla superficie del metallo uno strato protettivo costituito solo dall'ossido del componente legante. L'elemento di lega deve soddisfare i seguenti requisiti fondamentali: L'ossido che si forma sulla superficie del metallo deve essere continuo, cioè il rapporto Vok/VMe deve essere >1. Lo strato di ossido deve avere un'elevata resistenza ohmica rMeO > rFe, la dimensione degli ioni componenti la lega deve essere inferiore alla dimensione degli ioni del metallo base, vale a dire RM< rFe. Это условие вытекает из следующих соображений: 34 а) меньший радиус иона легирующего компонента по сравнению с ионом основного металла позволяет предполагать у легирующего компонента больший коэффициент диффузии в сплаве, что может обеспечить преимущественный выход ионов компонента на поверхность сплава и, следовательно, преимущественное образование оксида легирующего компонента; б) меньший радиус иона легирующего компонента позволит образовать оксид с соответствующими меньшими параметрами, который будет оказывать большое сопротивление диффузии через него и, следовательно, создавать большое затруднение для окисления основного металла. Это подтверждается сравнением размеров ионных радиусов железа и легирующих элементов: хрома, алюминия и кремния Ион металла Ионный радиус, Аº Fe2+ 0,74 Cr6+ 0,52 Al3+ 0,50 Si4+ 0,41. А энергия образования оксида легирующего компонента должна быть больше энергии образования оксида основного металла ΔGMeO<ΔGFeO. Если это условие не соблюдается, то оксид легирующего компонента будет восстанавливаться основным компонентом, как например, оксидная пленка меди, которая не может быть устойчивой на железном сплаве вследствие возможности протекания реакции CuO+Fe → FeO+Cu. При этом необходимо соблюдение условий: высокая температура плавления оксида легирующего компонента, низкая упругость диссоциации, а также отсутствие низкоплавких эвтектик в смеси с другими оксидами для того, чтобы оксид легирующего компонента был достаточно устойчив. В качестве примера можно привести элемент бор, который является аналогом Al по таблице Менделеева, но дает легкоплавкие оксиды. Температура плавления ВО3 − 294 0С, и поэтому бор, в отличие от алюминия, не может быть легирующим компонентом, повышающим жаростойкость. Необходимым условием является также, образование твердых растворов между легирующими компонентами и основным металлом, так как при этом условии возможно равномерное распределение этого элемента на поверхности и образование сплошной пленки оксида легирующего компонента. 35 Существует и другая теория, согласно которой легирующий элемент образует на поверхности сплава смешанные оксиды, обладающие повышенными защитными свойствами, по сравнению с оксидами чистых компонентов. Механизм повышения жаростойкости можно свести к тому, что легирующий компонент должен уменьшить возможность образования в окалине вюститной фазы (FeO) на стали и, таким образом, благоприятствовать образованию шпинельной фазы типа Fe3O4ּγFe2O3 с возможно меньшим параметром решетки. Жаростойкие легированные стали имеют на поверхности оксидные слои со структурой именно шпинели. Еще более высокими защитными свойствами обладают сложные шпинели типа FeOּMe2O3 или Fe2O3 ּMeO. С целью экономии легированных сталей жаростойкость углеродистых сталей повышают, применяя защитные покрытия. Для нанесения на сталь защитных покрытий пользуются следующими методами: ТЕРМОДИФФУЗИОННЫМ, НАПЛАВКОЙ И ПЛАКИРОВАНИЕМ. Для нанесения покрытий термодиффузионным методом стальное изделие после очистки поверхности механическим способом и травлением помещают в реакционную смесь, состоящую из порошка наносимого металла и катализатора. Например, при нанесении алюминиевых покрытий смесь содержит измельченный ферроалюминиевый и хлористый аммоний. Процесс насыщения поверхности стали алюминием (АЛИТИРОВАНИЕ) производится при высоких температурах (900−9500С), в результате чего происходит разложение хлористого аммония на аммиак и хлористый водород: NH4Cl → NH3 + HCl, который, в свою очередь, взаимодействует с алюминием 2Al +6HCl → 3H2 + 2AlCl3. На поверхности стального изделия происходит выделение атомарного алюминия AlCl3 +Fe → FeCl3 + Al, который диффундирует в сталь. Значительное повышение жаростойкости стали термодиффузионным покрытием обусловлено образованием окислов Al2O3 или двойных окислов FeAl2O4 в алитированном слое. На поверхности хромированных или силицированных изделий образуются соответствующие оксиды хрома или кремния. 36 НАПЛАВКА − это нанесение слоя металла или сплава на поверх- ность изделия посредством сварки плавлением. ПЛАКИРОВА́НИЕ − (фр. plaquer − накладывать, покрывать) термомеханическое покрытие − нанесение на поверхность металлических листов, плит, проволоки, труб тонкого слоя другого металла или сплава термомеханическим способом. Для защиты от карбонильной коррозии применяют хромистые стали с содержанием 30 % Сг, хромоникелевые стали с содержанием 23 % Сг и 20 % № и марганцевые бронзы доя работы при температуре до 700 °С и давлении до 35 МПа. При более низких параметрах возможно применение менее легированных сталей типа Х18Н9. Сырьем для синтеза мочевины CO(NH2)2 является NH3 и СО2. Процесс протекает при температуре 175−190°С и давлении 20 МПа. Хромистые нержавеющие стали различных марок непригодны для изготовления основных аппаратов. Наибольшую стойкость имеют стали, легированные молибденом, и хромникельмолибденовомедные стали. Важным фактором для повышения коррозионной устойчивости является тщательная очистка газов от сероводорода и дополнительное введение в систему кислорода в количестве 0,5- 1,0 об.% от содержания СО2. При сернистой коррозии сера и ее соединения - сернистый ангидрид (SO2), сероводород (H2S), меркаптаны или тиоспирты и т.д. являются достаточно агрессивными, коррозионно−активными веществами. Наиболее активным компонентом при высокотемпературной газовой коррозии является сероводород. Он даже более опасен, чем диоксид серы. Сернистый газ SO2 является исходным продуктом при производстве серной кислоты. Его получают при обжиге серного колчедана, сжигании серы, из сероводорода при утилизации отходящих газов металлургических производств. Чугунные детали скребков конверторных печей кипящего слоя, зубья и гребки колчеданных печей, котлы−утилизаторы, сухие электрофильтры, газоходы обжиговых газов в производстве серной кислоты часто выходят из строя вследствие газовой коррозии. В результате коррозии черных металлов в сернистом газе при температурах 300°С и выше образуется слоистая окалина, состоящая из FeS, FeO и др. 37 При температуре газа более 400 °С для деталей из чугуна характерно увеличение объема металла, достигающего 10 % от начальной величины. При этом резко снижается прочность материала. Детали испытывают коробление, трескаются и разрушаются. Это явление называется «ростом» чугуна и объясняется внутренним окислением металла. Максимальный рост чугуна наблюдается при 700 °С. К ростоустойчивым чугунам относятся высоколегированные хромистые чугуны, карбидный чугун типа «пирофераль» и «чугаль» Сернистый газ при высоких температурах окисляет никель. При этом образуется окалина, в состав которой входят NiS и NiO: 3Ni + S02 = NiS + 2NiO. Сернистый никель образуется и при действии на металл сероводорода: Ni + H2S = NiS + Н2. Сульфид никеля с металлическим никелем образует легкоплавкую эвтектику с температурой плавления около 625 °С. Образование этой эвтектики в сталях, содержащих никель, происходит преимущественно по границам зерен, вызывая разрушение металла. Стали с содержанием никеля выше 15 % очень чувствительны к действию сернистого газа. В процессе окисления они теряют механическую прочность. Поэтому при работе с газовой средой, содержащей диоксид серы, при температурах до 400 °С используют углеродистые стали, а при более высоких температурах − хромистые стали. Наиболее употребительны жаростойкие стали − 4Х9СА, Х6СЮ, XI7, ОХ 17 Г, X1800, Х25Т. Интенсивное образование окалины происходит при температурах выше 800−1000 °С. К жаропрочным сталям в этой среде относятся Х5М, Х6СМ, XI8Н12Т, Х23Н18. Рабочая температура для этих сплавов 550−600 °С (для Х23Н18 − 1000 °С). Сухой сернистый газ реагирует с алюминием очень медленно. Поэтому алюминий используют для защиты от коррозии деталей и узлов теплообменников и контактных аппаратов. Сухой сероводород при комнатной температуре не представляет опасности для обычных углеродистых сталей. С повышением температуры опасность сероводородной коррозии углеродистых сталей значительно увеличивается. При температуре выше 300 °С 38 железо подвергается сильной коррозии в серосодержащих газовых средах. Легирование хромом в количестве > Il 12% aumenta la resistenza alla corrosione a temperature fino a 700-800 °C. Quando gli acciai al cromo si corrodono, si formano incrostazioni, il cui strato esterno è costituito da solfuro di ferro. Non c'è praticamente cromo in questo strato. Tutto il cromo ossidato è concentrato nello strato interno, che ha proprietà protettive. I ferritici Fig. 5 10 15 20 hanno una buona resistenza chimica in un'atmosfera di zolfo-Cr,% in peso di idrogeno. 6. Tasso di corrosione delle leghe di cromo contenenti il 25–30% di acciaio solido in vapore d'olio a 650 °C. I numeri sulle curve indicano il cromo. Contenuto di H2S, % Particolarmente pericolosa è la presenza combinata di composti di zolfo e altri componenti corrosivi. Pertanto, nell'industria petrolifera, durante la lavorazione termica degli oli solforosi, una miscela di idrogeno solforato e idrogeno rappresenta un pericolo particolare. Da quelli mostrati in Fig. 6 dati mostrano che la velocità di corrosione degli acciai al cromo aumenta con l'aumentare della concentrazione di idrogeno solforato nei vapori d'olio. In questo caso, un aumento della concentrazione di H2S di 10 volte provoca un aumento della velocità di corrosione di oltre 12-15 volte. Quando il carburante brucia, si formano miscele di gas complesse contenenti O2 e vari ossidi, comprese le impurità di zolfo. In questi casi si osserva corrosione SOLFURO-OSSIDO. La pellicola protettiva sul metallo è solitamente composta da più strati. Lo strato esterno è arricchito con ossigeno ed è costituito da ossido metallico, mentre gli strati interni adiacenti alla superficie metallica contengono maggiori quantità di zolfo e solfuri. Se la combustione del carburante produce cenere, che contiene ossido di vanadio V2O5, la velocità di corrosione aumenta molto rapidamente. Le cause della corrosione degli acciai da vanadio sono state discusse in precedenza (vedere sezione 2.5.2). 39 Gli acciai al cromo contenenti 4−6% Cr sono considerati semiresistenti al calore. Gli acciai di questa classe, grazie alla loro disponibilità, maggiore resistenza alla corrosione e resistenza, sono ampiamente utilizzati nell'industria petrolifera per la produzione di unità di cracking. La resistenza al calore di questi acciai nell'aria e nei gas di scarico con un contenuto significativo di composti di zolfo a temperature di 500-600 ºС è circa 3 volte superiore alla resistenza al calore degli acciai non legati. 2.6.2. Metodi di protezione contro la corrosione da idrogeno Uno dei modi principali per aumentare la resistenza all'idrogeno degli acciai è quello di ALLEGArli con forti elementi che formano carburi che formano carburi più stabili della cementite. Tali elementi sono cromo, tungsteno, molibdeno, vanadio, niobio, titanio. Il tipo di fase del carburo ha una grande influenza sulla resistenza all'idrogeno dell'acciaio. Negli acciai legati al cromo, la resistenza all'idrogeno diminuisce a seconda della composizione della fase di carburo nelle seguenti serie: (Cr,Fe)23C6(Cr,Fe)23C6 + (Cr,Fe)7C3(Cr,Fe) 7C3 (Cr,Fe)7C3 + (Fe,Cr)3C(Fe,Cr)3C La serie sopra mostra che la massima resistenza all'idrogeno negli acciai al cromo si ottiene con la formazione di carburi del tipo (Cr,Fe )Tipo 23C6. La formazione di questo tipo di carburo con un contenuto di carbonio nell'acciaio dello 0,05% avviene quando nell'acciaio è presente almeno il 6% di cromo e con un aumento del contenuto di carbonio è necessario un aumento del contenuto di cromo. Quando si lega l'acciaio con elementi che formano carburi più forti del cromo, la massima resistenza all'idrogeno dell'acciaio si ottiene quando questi elementi sono contenuti in quantità sufficienti a legare tutto il carbonio nei carburi di tipo MeC. Nei casi in cui gli elementi dell'apparecchiatura operano in condizioni di elevati livelli di carichi meccanici, anche gli acciai resistenti all'idrogeno devono avere resistenza al calore. Per queste condizioni vengono utilizzati acciai con il 3% di Cr, ulteriormente legati con Mo, V o W, ad esempio acciaio di qualità 20Х3МВФ (EI579). I gradi di acciaio X5M, X5VF, X9M, 1X13 hanno una resistenza ancora maggiore. Nelle condizioni più severe vengono utilizzati gli acciai al cromo-nichel resistenti al calore 0Х18Н10Т o Х17Н17М2Т. 40 Un altro metodo ampiamente utilizzato per aumentare la resistenza all'idrogeno degli acciai è il RIVESTIMENTO con metalli che hanno una permeabilità all'idrogeno inferiore rispetto al metallo base. Ciò consente di ridurre la concentrazione di idrogeno nell'interfaccia tra il metallo base e lo strato di rivestimento e di ridurre il suo effetto aggressivo sul metallo base. Il calcolo della pressione effettiva dell'idrogeno all'interfaccia del metallo del rivestimento di base viene effettuato secondo l'equazione: 1/2 1/2 p2 = p1 (11) + dove p1 è la pressione dell'idrogeno sulla superficie esterna del rivestimento strato di rivestimento; p2 è la pressione effettiva dell'idrogeno all'interfaccia del rivestimento-metallo di base; = VН1/VН2 è il rapporto tra la permeabilità all'idrogeno dello strato di rivestimento e del metallo base, rispettivamente; = l1/l2 – rapporto rispettivamente tra lo spessore dello strato di rivestimento e del metallo base. Per aumentare la permeabilità all'idrogeno, i metalli e le leghe possono essere disposti in fila: alluminio, rame, nichel, X18N10T, 0X13. In pratica, le industrie chimiche e di raffinazione del petrolio utilizzano principalmente bimetalli realizzati con acciai al carbonio o bassolegati con uno strato protettivo di acciai 0Х13 o Х18Н10Т. La temperatura operativa delle apparecchiature realizzate con l'acciaio selezionato deve essere mantenuta a 25°C al di sotto della temperatura alla quale è possibile la corrosione da idrogeno. Capitolo 3. CORROSIONE ELETTROCHIMICA DEI METALLI 3.1. Potenziali degli elettrodi dei metalli Quando un metallo è immerso in un elettrolita, si verifica un salto di potenziale al confine di fase dovuto alla formazione di un doppio strato elettrico 5, 6. Se una piastra metallica, ad esempio una piastra di rame, viene immersa in acqua o in una soluzione di sale di rame, gli ioni Cu2+ caricati positivamente inizieranno a spostarsi dallo strato metallico situato al confine con l'acqua nell'acqua. In questo caso, nel reticolo cristallino del metallo appare un eccesso di elettroni e la piastra acquisisce una carica negativa. Tra la piastra caricata negativamente e gli ioni positivi che sono passati nella soluzione si crea un'attrazione elettrostatica, che impedisce l'ulteriore transizione degli ioni rame nella soluzione, cioè il processo di dissoluzione del metallo si interrompe. Allo stesso tempo, si sviluppa il processo opposto: gli ioni di rame dalla soluzione, avvicinandosi alla superficie della piastra, accettano elettroni da essa e passano in uno stato neutro. Dopo un certo periodo di tempo si stabilisce uno stato di equilibrio dinamico, in cui la velocità di transizione degli ioni dal metallo alla soluzione è uguale alla velocità di scarico degli ioni dalla soluzione al metallo. Il fenomeno descritto è rappresentato schematicamente in Fig. 7 (gli ioni metallici sono mostrati non idratati per semplicità). L'equilibrio tra gli ioni nella soluzione e il metallo per l'esempio in esame è espresso dall'equazione: Cu2+р−р + 2ē Cuºcristallo Nell'equazione di equilibrio di una reazione elettrochimica, gli elettroni sono solitamente scritti sul lato sinistro, vale a dire, il processo di riduzione viene svalutato. Fig.7. Schema dell'aspetto di un doppio strato elettrico all'interfaccia metallo-soluzione Come si può vedere dall'esempio sopra, quando un metallo entra in contatto con una soluzione del suo sale, le due fasi in contatto acquisiscono cariche opposte. Di conseguenza, sull'interfaccia di fase si forma un doppio strato elettrico e si verifica un salto di potenziale () tra il metallo e la soluzione. Se si immergono nella soluzione due piastre di metalli diversi (M1 e M11), si creerà una coppia galvanica in cui il metallo meno attivo verrà ridotto e il metallo più attivo verrà ossidato (Fig. 8). Riso. 8. Schema di funzionamento di una cella galvanica In generale, il lavoro di questa coppia galvanica è determinato dalla differenza di potenziale (fem della cella galvanica) ed è accompagnato dal bilancio delle semireazioni: E = ox − rosso, dove ox è il potenziale dell'ossidante, V; rosso – potenziale dell'agente riducente, V. Men+(ox) + ne ⇆ Меº Meº(rosso) − ne ⇄ Меn+. Quando il numero di cationi che passano in soluzione nell'unità di tempo diventa uguale al numero di cationi depositati sulla superficie del metallo, si verificherà un equilibrio dinamico e il processo di dissoluzione si fermerà. Pertanto, la transizione di un gran numero di atomi di ioni metallici in soluzione in tali condizioni è impossibile. Tuttavia, se l'equilibrio del doppio strato elettrico viene disturbato dalla scarica di elettroni o dalla rimozione di atomi di ioni metallici, il processo di corrosione procederà senza ostacoli. I potenziali degli elettrodi dei metalli che sono in equilibrio con i propri ioni sono chiamati equilibrio. Il valore del potenziale di equilibrio può essere calcolato per qualsiasi attività ionica utilizzando l'equazione di Nernst: E = Eº + (RT /nF)ln(aMen+), (12) n+ dove Eº è la differenza di potenziale standard ad aMe = 1; R – costante universale dei gas, 8,3 kJ/kmolK; T – temperatura assoluta, ºK; n – valenza metallica; F – Numero di Faraday 96500 C/mol; 43 aМen+ – attività degli ioni metallici in mol/l. Se sostituiamo tutte le costanti a 25°C (T = 298 K) e moltiplichiamo per 2,3 per passare dai logaritmi naturali a quelli decimali, otteniamo la seguente espressione E = Eº + (0,0592/n)log(aMen+), ( 13) Quando la soluzione viene diluita, il potenziale del metallo si sposta verso il lato negativo. Se, ad esempio, l’attività degli ioni Zn2+ in una soluzione di sale di zinco è 10–2 mol/l, allora il potenziale di equilibrio dello zinco immerso in questa soluzione sarà pari a: E = − 0,76 + (0,0592/2) lg10−2 = − 0,819 V, (14) Con un aumento della concentrazione di ioni metallici nella soluzione, il potenziale metallico, al contrario, si sposta nella direzione positiva. Pertanto, se l'attività degli ioni zinco viene considerata pari a 10 mol/l, allora il potenziale dello zinco sarà E = − 0,76 + (0,0592/2)log10 = − 0,73 V, (15) Potenziali degli elettrodi dei metalli in cui L' Il processo di scambio che determina il potenziale coinvolge non solo i propri, ma anche altri ioni e atomi, che sono chiamati non equilibrio o irreversibili. Per i potenziali di non equilibrio la formula di Nernst non è applicabile, poiché le reazioni che avvengono sul metallo, ad es. la perdita e l'acquisizione di elettroni avvengono in modi diversi e il potenziale non può caratterizzare l'inizio dell'equilibrio di nessuna reazione sull'elettrodo. L'entità dei potenziali di non equilibrio è influenzata dalla natura dell'elettrolita, dalla temperatura, dal movimento dell'elettrolita, dalla concentrazione della soluzione, ecc. Ad esempio, il potenziale dell'alluminio in NaCl al 3% è − 0,6 V, in Na2SO4 0,05 M − 0,47 V, e in 0,05 M Na2SO4 + H2S − 0,23 V. A causa dell'eterogeneità elettrochimica della superficie del metallo o dell'elettrolita, sul metallo si formano aree con potenziali diversi. Le ragioni principali che causano l'eterogeneità elettrochimica di una superficie metallica possono essere le seguenti: − LIQUAZIONE − (La liquation, Saigerung) - è la capacità delle leghe di disintegrarsi durante la transizione dallo stato liquido a quello solido in componenti o singoli composti che hanno punti di fusione diversi ; − eterogeneità del film protettivo; 44 – eterogeneità delle condizioni fisiche – temperatura disuguale nelle diverse zone del metallo, presenza di aree deformate, concentratori di stress; − eterogeneità dell'elettrolita - differenza nella concentrazione di ossigeno o concentrazione di sale, diverso valore di pH. Le zone con potenziale più negativo sono dette ANODICHE, le zone con potenziale più positivo sono dette CATODO. Il grado di eterogeneità della superficie metallica è determinato dalla differenza di potenziale delle sezioni catodica e anodica. 3.2. Termodinamica della corrosione elettrochimica La possibilità del verificarsi spontaneo del processo di corrosione è determinata dalla diminuzione dell'energia libera (potenziale isobarico-isotermico) G< 0: G = −nFE < 0, (16) где n – эквивалентное число электронов; Е = К – А – эдс гальванического элемента, образующегося при контакте металла с окислителем в процессе коррозии; К – обратимый потенциал катодной реакции (окислителя); А – обратимый потенциал анодной реакции (металла – восстановителя); F – число Фарадея. Принципиальная возможность протекания процесса электрохимической коррозии металла имеет место при К > A o E > 0. Il potenziale redox reversibile dell'agente ossidante nell'elettrolita deve essere più positivo in queste condizioni rispetto al potenziale reversibile del metallo nelle stesse condizioni. Per stabilire i limiti della possibilità termodinamica della corrosione elettrochimica dei metalli si possono utilizzare i diagrammi di Pourbaix. I diagrammi sono rappresentazioni grafiche della dipendenza dei potenziali degli elettrodi reversibili (in volt sulla scala dell'idrogeno) dal pH della soluzione per gli stati di equilibrio: con la partecipazione di elettroni - linee orizzontali; con la partecipazione di elettroni e ioni H+ o OH– - linee inclinate; con la partecipazione di ioni H+ e OH–, ma senza la partecipazione di elettroni (valore del pH della formazione di idrati) – linee verticali. 45Fig.9. Diagramma E – pH per il sistema Al-H2O a 25°C Come esempio in Fig. La Figura 9 mostra il diagramma di Pourbaix per il sistema Al – H2O. Ciascuna regione del diagramma corrisponde ad uno stato termodinamicamente stabile: lo stato metallico, sotto forma di ioni in soluzione, sotto forma di ossidi o idrossidi. Poiché il raggiungimento dell'equilibrio in una soluzione dipende non solo dagli ioni idrogeno, ma anche da altri ioni, la delimitazione delle regioni è formata da diverse linee di equilibrio. Ognuna di queste linee corrisponde ad un'attività specifica degli ioni corrispondenti. Nella regione nella parte inferiore del diagramma, l'alluminio metallico è termodinamicamente stabile e non si corrode. Nella parte sinistra sopra la linea orizzontale, lo stato dell'alluminio sotto forma di ioni Al3+ in soluzione è termodinamicamente stabile. La regione centrale corrisponde alla stabilità delle fasi solide dell'ossido Al2O3 o dell'idrossido Al(OH)3, che formano una pellicola protettiva sulla superficie dell'alluminio durante il processo di corrosione. Il lato destro caratterizza le condizioni in cui l'alluminio subirà corrosione con la formazione dell'anione AlO2– in soluzione. 3.3. Meccanismo di corrosione elettrochimica La corrosione dei metalli negli elettroliti avviene con la formazione di coppie galvaniche, chiamate coppie di corrosione. Se si abbassano rispettivamente le piastre di zinco e di ferro in un recipiente con soluzioni di cloruri di zinco e ferro (simile al diagramma pH me in Fig. 8) e le si collega con un conduttore esterno, un milliamperometro collegato al circuito mostrerà il presenza di corrente elettrica nel circuito. Si forma una coppia galvanica nella quale lo zinco si ossida e va in soluzione (anodo). Il ferro non si ossida e non entra in soluzione poiché, essendo combinato con lo zinco, agisce come un catodo. Il ferro, non combinato con lo zinco, si corrode in una soluzione simile. In pratica, le leghe contengono un gran numero di microelementi e metalli dissimili. In altre parole, metalli diversi si trovano sullo stesso piano nel contatto diretto tra loro e con l'elettrolita. Il risultato è una cella multielettrodica in cui gli anodi si alternano ai catodi. Nei siti anodici gli ioni metallici (Men+) vanno in soluzione. Gli elettroni rilasciati (nē) si muovono attraverso il metallo dall'anodo al catodo. La dissoluzione degli ioni metallici in soluzioni acquose dovrebbe essere rappresentata come il risultato dell'interazione degli ioni - atomi situati sulla superficie esterna del metallo con molecole d'acqua polari. L'interazione degli ioni metallici disciolti con i dipoli d'acqua è causata dalle forze di attrazione elettrostatica. Di conseguenza, attorno a ciascun ione, in misura maggiore o minore (a seconda dell'entità della sua carica), si forma un guscio di dipoli d'acqua (fenomeno di idratazione). A causa dell'idratazione, il raggio effettivo dello ione aumenta, di conseguenza la mobilità degli ioni idratati diminuisce in modo significativo. Il processo di idratazione è accompagnato dal rilascio di energia, mentre il processo di disidratazione richiede energia. Oltre ai dipoli d'acqua, lo ione può essere ricoperto da un guscio di altri dipoli. Più in generale, questo fenomeno è chiamato solvatazione15. Pertanto, la corrosione elettrochimica consiste delle seguenti fasi, che si verificano in parallelo: - un processo anodico, che consiste nella transizione degli ioni metallici dalla superficie nella soluzione e nella loro idratazione: SOLVAZIONE - il processo di formazione di associati tra un solvente e una soluzione disciolta sostanza, il prodotto della solvatazione è SOLVATO. Se il solvente è l'acqua, allora IDRATAZIONE, il prodotto dell'idratazione è IDRATO. 15 47 Meº − nē Uomini+ + mH2O Uomini+ mH2O. l'idrato è un processo catodico, che consiste nell'assimilazione (cattura) di elettroni da parte di un depolarizzatore (D): D + nē . Poiché i processi anodico e catodico sono indipendenti e si verificano più facilmente: l'anodico in aree con un potenziale iniziale della superficie più negativo e il catodico in quelle più positive - e in condizioni pratiche l'eterogeneità elettrochimica della superficie metallica necessaria per questo è notato, questi processi si verificano prevalentemente in modo localizzato. Il flusso di corrente elettrica viene effettuato sul metallo - dal movimento degli elettroni dalle aree anodiche al catodo e nella soluzione, allo stesso tempo, avviene il movimento degli ioni. Pertanto, l'intensità della corrente può e serve come criterio per la velocità di corrosione elettrochimica. Calcolare il consumo di materiale dell'anodo, ad es. la quantità di metallo corroso da 1 cm2 della sua superficie può essere utilizzata utilizzando la formula di Faraday: K = Q A / F n = i A / F n, dove K è la quantità di metallo corroso metallo, g/cm2 ; Q è la quantità di elettricità che scorre nel tempo τ, [s] tra le sezioni anodica e catodica; i – densità di corrente, A/cm2; F – Numero di Faraday; n – valenza metallica; A è la massa atomica del metallo. 3.4. Polarizzazione dei processi degli elettrodi e sue cause Quando gli elettrodi di un elemento di corrosione vengono cortocircuitati, si verifica un cambiamento significativo nei potenziali degli elettrodi, ma la resistenza della coppia praticamente non cambia. In questo caso si osserva una diminuzione della forza elettromotrice dell'elemento corrosivo. Una variazione dei potenziali iniziali, che porta ad una diminuzione della corrente di corrosione e, di conseguenza, della velocità di corrosione, è chiamata POLARIZZAZIONE. La variazione dei potenziali (16) degli elettrodi durante il funzionamento dell'elemento corrosivo mostra che il potenziale del catodo diventa più negativo (POLARIZZAZIONE CATODICA) e il potenziale dell'anodo diventa più positivo (POLARIZZAZIONE ANODICA): K = Ko – K, (17) o A = A + A, (18) dove K e A sono i valori potenziali degli elettrodi dell'elemento di lavoro; Кº e Аo – valori iniziali dei potenziali degli elettrodi prima della chiusura del circuito; K e A – spostamento potenziale (polarizzazione) del catodo e dell'anodo. Pertanto la differenza di potenziale E = K − A diminuisce. LA POLARIZZAZIONE è una conseguenza del ritardo dei processi dell'elettrodo dal flusso di elettroni in un elemento cortocircuitato. Le resistenze ohmiche (R) hanno scarso effetto sulla riduzione della corrente di corrosione, poiché solitamente sono piccole. Di grande importanza sono le resistenze di polarizzazione, che sono associate sia a difficoltà nella scarica di elettroni al catodo (Pk), sia a difficoltà nella transizione degli ioni metallici positivi dal reticolo metallico alla soluzione (PA). Queste resistenze, che hanno dimensione ohmica, sono la ragione principale della diminuzione della velocità di corrosione elettrochimica. L'intensità della corrente al momento della chiusura del circuito secondo la legge di Ohm è determinata dalla formula Iinit = (Кº − Аo)/R, (19). Il valore iniziale della corrente di corrosione (Iinit) è direttamente proporzionale alla differenza tra i valori di potenziale iniziale Kº e Ao, purché Kº > Ao, ed inversamente proporzionale alla resistenza. La corrente di corrosione stazionaria (Icella di lavoro) è determinata dall'equazione: Icella di lavoro = (K – A)/(R + PA + PK). Quindi lavoro. el−ta< Iнач. Снижению скорости электрохимической коррозии или уменьшению коррозионного тока могут способствовать: − уменьшение степени термодинамической стабильности, т. е. сближение равновесных потенциалов анодного Аº и катодного Кº процессов; − торможение катодных процессов, иначе говоря, увеличение катодной поляризуемости РК; 49 − торможение анодных процессов, то есть увеличение анодной поляризуемости РА. Поляризация является весьма желательным явлением в коррозионных процессах, так как электроды замкнуты накоротко, омическое сопротивление их невелико и, следовательно, не будь поляризационных сопротивлений, коррозия протекла бы весьма интенсивно. Всякое воздействие, уменьшающее поляризацию, увеличивает скорость коррозионных процессов. Факторы, уменьшающие поляризацию электродов коррозионного элемента, называют ДЕПОЛЯРИЗАТОРАМИ, а процессы, протекающие при этом – АНОДНОЙ И КАТОДНОЙ ДЕПОЛЯРИЗАЦИЕЙ. 3.4.1. Анодная поляризация Анодная поляризация обычно оказывает меньшее влияние на изменение разности потенциалов элемента по сравнению с катодной поляризацией. Анодный процесс, заключающийся в ионизации металла и гидратировании ионов по уравнению: Men+ + mH2O → Men+ ∙ mH2O, может протекать беспрепятственно только при условии беспрерывного отвода образующихся ионов из прианодной зоны. Торможение анодного процесса будет иметь место и в случае отставания скорости выхода ионов металла в раствор от скорости перетекания электронов на катодные участки. В результате анодной поляризации отрицательный заряд на металлической обкладке двойного слоя уменьшается, а следовательно, потенциал металла сдвинется в положительную сторону (или станет менее отрицательны). Таким образом, анодная поляризация происходит или в связи с тем, что скорость анодного процесса не поспевает за скоростью отвода электронов (перенапряжение ионизации металла), или потому, что из−за недостаточно быстрого отвода перешедших в раствор ионов металла повышается концентрация этих ионов в прианодной зоне (концентрационная поляризация). Чем легче протекает анодный процесс, тем меньше его поляризуемость. Следить за тем, легко ли протекает анодный процесс или он заметно тормозится, можно по ходу поляризационной анодной кри50 Потенциал, В вой. Эта кривая показывает зависимость потенциала электрода от плотности анодного тока на нем. Такая кривая показана на рис. 10. Удаление продуктов анодной реакции вызывает деполяризацию анода. Деполяризация анода может быть ускорена перемешиЕ Е ванием раствора, образованием о А таких продуктов коррозии, которые выпадают в осадок, а также путем образования комплексных соединений. Вследствие связывания в комплекс ионов металла, перешедших в раствор, концентрация их в прианодной зоне уменьшаПлотность тока, мА/см2 ется; анодная поляризация сниРис. 10. Анодная поляризаци- жается и переход ионов в расонная кривая твор, т.е. коррозия, усиливается. Например, медь и ее сплавы образуют в растворе аммиака хорошо растворимый комплексный ион 2+. Этим объясняется сильная коррозия меди и ее сплавов в аммиачных растворах. Поляризация анодного процесса в обычных условиях происходит в незначительной степени. Исключение представляет случай возникновения анодной пассивности. 3.4.2. Катодная поляризация Протекание катодной реакции могут тормозить два основных процесса: − торможение за счет трудности протекания самой реакции присоединения электрона деполяризатором, т. е. задержка в реакции nē + D (Dnē). Затруднение протекания катодного процесса по этой причине называется ПЕРЕНАПРЯЖЕНИЕМ РЕАКЦИИ КАТОДНОЙ ДЕПОЛЯРИЗАЦИИ; − торможение в процессе подвода к катодной поверхности деполяризатора (D) или отвода от катодной поверхности продуктов восстановления деполяризатора (Dnē). Затруднение катодного процесса по этой причине называется КОНЦЕНТРАЦИОННОЙ ПОЛЯРИЗАЦИЕЙ КАТОДА. 51 Всякий процесс захвата электрона на катоде, т.е. всякий процесс восстановления какого−либо вещества может являться катодной деполяризующей реакцией. Деполяризация катода может протекать различными путями: − ассимиляция электронов ионами: 2H+ +2ē H2, Fe3+ + ē Fe2+, Cu2+ + ē Cu+, S2O8– + ē 2SO42–; − ассимиляция электронов нейтральными молекулами: О2 + 4ē + 2Н2О 4ОН–, Cl2 + 2ē 2Cl–, Н2О2 + 2ē 2ОН–; − ассимиляция электронов нерастворимыми пленками Fe3O4 + 2ē + H2O 3FeO + 2OH–, Fe(OH)3 + ē Fe(OH)2 + OH–; − ассимиляция электронов органическими соединениями RO + 2ē + 4H+ RH2 + H2O, R + 2ē + 2H+ RH2, где R – радикал или органическая молекула. Наиболее часто встречаемыми и наиболее важными катодными деполяризационными процессами являются два: а) разряд и выделение водорода (деполяризатор ион водорода) 2H+ +2ē H2. В этом случае коррозионный процесс называется коррозией с ВОДОРОДНОЙ ДЕПОЛЯРИЗАЦИЕЙ. Этому виду коррозии подвержены электроотрицательные металлы в кислотах и весьма электроотрицательные металлы, например Mg, в воде и нейтральных растворах электролитов; б) восстановление атомов или молекул кислорода электронами, притекающими на катодных участках − КИСЛОРОДНАЯ ДЕПОЛЯРИЗАЦИЯ: О2 + Н2О + 4ē 4ОН–. Деполяризатором здесь является растворенный в электролите кислород. Этот вид коррозии является наиболее распространенным. Так корродирует большинство металлов в атмосфере, почве, нейтральных растворах электролитов. Иногда оба деполяризующие процесса протекают параллельно, как например, при коррозии железа в разбавленных растворах серной кислоты в присутствии кислорода воздуха. 52 3.4.3. Водородная деполяризация Протекание коррозии металла с водородной деполяризацией возможно, если (Me)обр < (н2)обр. (20) Согласно уравнению Нернста (н2)обр = (н2)ºобр + (RT2,3/F)lg(aH+/pH21/2), (21) о где (н2) обр – стандартный обратимый потенциал водородного электрода при всех температурах (при ан+ = 1 и рН2 = 1 ат); R – универсальная газовая постоянная. Ниже приведены значения обратимого потенциала водородного электрода (н2)обр при 25 оС и различных значениях рН среды и pН2. рн2, ат 5∙10 1 –7 0 + 0,186 0 рН среды 7 − 0,228 − 0,414 14 − 0,641 − 0,828 Изменение потенциала катода при катодной деполяризации выделением водорода подчиняется ближе всего логарифмической зависимости от плотности тока. Эта зависимость определяется выражением: К = Кº – (а + b lg i). (22) Для очень малых плотностей катодного тока (меньше 10–4 – 10–5 А/см2) потенциал от плотности тока имеет линейную зависимость. Поэтому при i = 0 К = Кº, а не бесконечности, как это следует из уравнения. Поляризуемость катода при выделении на нем водорода принято определять ВЕЛИЧИНОЙ ПЕРЕНАПРЯЖЕНИЯ ВОДОРОДА на данном материале катода. Под ПЕРЕНАПРЯЖЕНИЕМ ВОДОРОДА понимают сдвиг потенциала катода при данной плотности тока в отрицательную сторону по сравнению с потенциалом водорода в том же растворе (без наложения тока) и обозначают через η η = − (К − Кº) (23) или η = a + blgI, (24) где b – константа, не зависящая от материала, рассчитывается как 2,3 ∙ 2RT / nF; а − константа, зависит от природы и состояния поверхности материала катода и численно равна величине перенапряжения при плотности тока, равной единице. 53 Поляризационная кривая для случая водородной деполяризации имеет вид кривой Кº MN (рис. 11). − N 1 M η Рис. 11. Катодная поляризационная кривая для процесса водородной деполяризации ко или (н2)обр i1 0 i P Если по оси абсцисс взять не плотность тока, а логарифм этой величины, то кривая перенапряжения будет прямой. В этом случае константа b является тангенсом угла наклона полученной прямой к оси абсцисс, константа а равна величине ординаты lg i = 0 (т.е. при плотности тока i = 1). Величина перенапряжения водорода зависит от плотности тока, т.е. при данной силе тока, от истинных размеров поверхности катода. При одинаковой силе тока она больше на гладкой поверхности, чем на шероховатой. 3.4.4. Кислородная деполяризация Процессы коррозии с КИСЛОРОДНОЙ ДЕПОЛЯРИЗАЦИЕЙ могут протекать в том случае, если при данных условиях обратимый потенциал металла отрицательнее обратимого потенциала кислородного электрода (Me)обр < (О2)обр. (25) 54 Обратимый потенциал кислородного электрода может быть вычислен по уравнению Нернста: (О2)обр = (О2)ºобр + (RT2,3/4F)lg(pО2/a4ОH−) (26) где (О2)ºобр – стандартный обратимый потенциал кислородного электрода (при аOH– = 1 и рО2 = 1 ат); рО2 – парциальное давление кислорода; аОН– – активность гидроксильных ионов. Величина обратимого потенциала кислородного электрода (О2)ºобр при 25 oC для различных значений рН и рО2: рН среды ро2 , ат 0,21 1 0 + 1,218 + 1,229 7 + 0,805 + 0,815 14 + 0,391 + 0,400 Коррозия с кислородной деполяризацией является термодинамически более возможным процессом, чем коррозия с водородной деполяризацией, так как равновесный потенциал восстановления кислорода более положителен, чем равновесный потенциал выделения водорода. Коррозия металлов с кислородной деполяризацией является самым распространенным коррозионным процессом. Коррозии с кислородной деполяризацией подвергаются металлическая обшивка нефтезаводской аппаратуры и оборудования, детали аппаратов, соприкасающиеся с водой и нейтральными водными растворами солей (например, трубы конденсационно−холодильного оборудования), подземные трубопроводы, днища резервуаров и др. При коррозии металлов с кислородной деполяризацией катодный процесс определяется перенапряжением катодной реакции и скоростью подвода кислорода к электроду. Замедленность катодной реакции по схеме: О2 + 2Н2О + 4е 4ОН– (в щелочных или нейтральных растворах), или по схеме: О2 + 4Н+ + 4е 2Н2О (в слабо−кислых растворах) называется ПЕРЕНАПРЯЖЕНИЕМ ИОНИЗАЦИИ КИСЛОРОДА. При больших плотностях тока (при iк > 10–2 A/m2) e un tasso significativo di fornitura di ossigeno al catodo, la sovratensione di ionizzazione dell'ossigeno ha una dipendenza logaritmica dalla densità di corrente 55 δ" = а"+ b" log i, (27) dove δ" è la sovratensione della ionizzazione dell'ossigeno; a" è una costante che dipende dal materiale del catodo e dallo stato della sua superficie, dalla temperatura e da altri fattori. È definita numericamente come il valore di sovratensione a i = 1; b" è una costante che non dipende dal materiale del catodo; si determina dall'uguaglianza b" = 2RT/nF ∙ 2,3; alla temperatura di 20° e n = 1, b" = 0,117. Il meccanismo di riduzione catodica dell'ossigeno è piuttosto complesso e avviene in più fasi: dissoluzione dell'ossigeno nell'elettrolita, trasferimento dell'ossigeno disciolto alle sezioni catodiche del metallo, ionizzazione dell'ossigeno. A differenza della depolarizzazione dell'idrogeno, per la quale la polarizzazione della concentrazione è praticamente insignificante, nella corrosione dei metalli con depolarizzazione dell'ossigeno essa gioca un ruolo molto importante a causa della velocità limitata di apporto di ossigeno al catodo. Ciò è spiegato dalla bassa solubilità dell'ossigeno nella soluzione elettrolitica e, di conseguenza, dalla sua bassa concentrazione, dalla difficoltà di diffusione dell'ossigeno attraverso uno strato stazionario di liquido adiacente al catodo. Con forte agitazione della soluzione, circolazione intensa o in condizioni che forniscono una significativa aerazione dell'elettrolita, il processo di corrosione viene inibito da una sovratensione della ionizzazione dell'ossigeno. I processi di diffusione influenzano la velocità di corrosione quando il metallo è immerso in una soluzione calma o leggermente agitata. La curva di polarizzazione catodica generale ha una forma complessa (Fig. 12) ed è la somma di tre curve che caratterizzano la polarizzazione durante la ionizzazione dell'ossigeno (AB), la polarizzazione della concentrazione (DE) e la scarica degli ioni idrogeno (GFH). Nella prima sezione, la velocità del processo catodico, cioè la quantità di ossigeno ridotta per unità di tempo è inferiore alla velocità massima possibile di apporto di ossigeno alla superficie per diffusione. A questo proposito, in quest'area la velocità del processo è limitata principalmente dalla lentezza del processo di riduzione dell'ossigeno stesso. 56 − H F K E G (H2)arr 0 (O2)arr A D B C iä i Fig. 12 Curva di polarizzazione catodica per il processo di depolarizzazione dell'ossigeno Con un ulteriore aumento della densità di corrente, il potenziale si sposta nella direzione negativa, dapprima gradualmente, e poi bruscamente (sezione CDE). Un brusco spostamento del potenziale nella direzione negativa è associato alla polarizzazione della concentrazione dovuta a una diminuzione della concentrazione di ossigeno. Ad un certo valore potenziale diventa possibile un nuovo processo catodico, solitamente associato alla depolarizzazione dell'idrogeno (sezione EK). 3.5. Controllo del fattore di corrosione Il processo di corrosione elettrochimica dei metalli e delle leghe è costituito da processi o fasi elementari. La velocità effettiva del processo di corrosione dipende direttamente dalla sua totale inibizione in ogni fase. La percentuale di inibizione dell'intero processo in ciascuna delle sue fasi caratterizza il GRADO DI CONTROLLO DEL PROCESSO in una determinata fase. 57 è chiamato lo stadio che determina la velocità di corrosione, poiché ha la maggiore resistenza rispetto agli altri stadi. PROCESSO DI CONTROLLO o2)arr imax I imax b b b a (o2)arr in A A imax (Me)arr (Me)arr K ∆K R P (o2)arr imax I I S (о2)arr i I g g d d Fig. 13. Diagrammi di corrosione di polarizzazione per i principali casi pratici di monitoraggio dei processi di corrosione elettrochimica 58 I in In condizioni pratiche, esistono diversi casi base di monitoraggio della corrosione elettrochimica di metalli e leghe, comprovati da N.D. Tomashov. Bekkerev I.V. "Metalli e leghe - Gradi, composizione chimica", Parte 2, Ulyanovsk: Università tecnica statale di Ulyanovsk, 2007. - p. 630, http://www.bibliotekar.ru/spravochnik-73/index.htm. 3]. Semenova I.V. Corrosione e protezione dalla corrosione, M.: Fizmatlit, 2002, 335 p. 4]. Khaldeev G.V., Borisova T.F. “Permeabilità all'idrogeno di metalli e leghe nei processi elettrochimici di corrosione. Elettrochimica" - Volume 30, Mosca, 1989 p. 232 5]. T. L. Lukanina, T. T. Ovchinnikova, V. Ya Sigaev, "Chimica generale" Parte II. Esercitazione. 2a edizione, corretta e ampliata, 2006)