Odporność chemiczna materiałów budowlanych w zależności od ich składu i struktury. Właściwości technologiczne materiałów. Odporność chemiczna materiałów Władysław Aleksandrowicz Lichaczow x Główny kierunek pracy
Ministerstwo Szkolnictwa Ogólnego i Zawodowego
Federacja Rosyjska
MOSKWA PAŃSTWOWY UNIWERSYTET INŻYNIERII ŚRODOWISKA
A.G.Parshin V.S.Pakhomov D.L.Lebedev
Odporność chemiczna materiałów i ochrona antykorozyjna
Warsztat laboratoryjny
Pod redakcją dr Tech. Nauki A.A. Szewczenko
Moskwa-1998
BBK35.11 Χ 46
Recenzenci:
Katedra Korozji Moskiewskiej Państwowej Akademii Ropy i Gazu im. Gubkin;
Doktorat technologia Sciences A.S. Abramow, firma zajmująca się ochroną środowiska „ShanEco”, Moskwa.
Zatwierdzony jako pomoc dydaktyczna przez radę redakcyjną i wydawniczą Moskiewskiego Państwowego Uniwersytetu Inżynierii Środowiska.
Parshin A.G., Pakhomov V.S., Lebedev D.L.
Χ 46 Odporność chemiczna materiałów i ochrona antykorozyjna: Warsztat laboratoryjny / wyd. A.A. Szewczenko – M.: MSUIE, 1998.-80 s.; chory.8.
ISBN 5-230-11142-9
W ramach pracowni znajdują się cztery prace laboratoryjne dotyczące korozji elektrochemicznej i ochrony metali: „Wpływ niejednorodności strukturalnej na korozję metali podczas redukcji jonów wodorowych”, „Korozja metali z depolaryzacją tlenową”, „Potencjały elektrod”, „Korozja kontaktowa i katodowej ochrony metali przed korozją”. Zarysowano podstawowe pojęcia teorii korozji elektrochemicznej metali oraz metodykę prowadzenia badań korozyjnych. Przeznaczony jest dla studentów III i IV roku studiów stacjonarnych, studiujących kierunki odporności chemicznej materiałów i ochrony antykorozyjnej.
ISBN 5-230-11142-9 UDC620.193 BBK 35.11
© A.G.Parshin, V.S.Pakhomov, D.L.Lebedev.1998
© MSUIE, 1998
Przedmowa
Warsztaty laboratoryjne są napisane zgodnie z programem zajęć „Odporność chemiczna materiałów i ochrona antykorozyjna”, przewidzianym w programie nauczania szeregu specjalności. Warsztaty obejmowały prace dotyczące korozji elektrochemicznej metali, które zostały opracowane w Katedrze Materiałów Kompozytowych i Zabezpieczeń przed Korozją.
Każda praca laboratoryjna rozpoczyna się od części teoretycznej. Omówiono mechanizm, termodynamikę i kinetykę korozji podczas katodowej redukcji jonów wodorowych, depolaryzację tlenu, procesy równowagowe i nierównowagowe na granicy faz metal-elektrolit oraz teorię potencjałów elektrodowych.
Przy przedstawianiu zagadnień teoretycznych posłużono się nowoczesnymi koncepcjami elektrochemicznymi dotyczącymi mechanizmu i kinetyki procesów korozyjnych.
Wykonywanie prac laboratoryjnych pozwala studentom lepiej zrozumieć podstawy badań nad korozją i ochroną metali, a także wpaja umiejętności prowadzenia podstawowych laboratoryjnych badań korozyjno-elektrochemicznych.
Prowadzenie dziennika laboratoryjnego
1) tytuł i cel utworu;
2) schemat instalacji;
3) tabelę z wynikami doświadczeń i ich przetwarzaniem (obliczenia, wykresy);
4. Konkluzje.
Dziennik należy prowadzić starannie i niezwłocznie oczyścić, tj. w taki sam sposób, w jaki prowadzona jest dokumentacja wszystkich działań badawczych i testowych.
Schemat instalacji powinien być jasny i zrozumiały, bez ustnych wyjaśnień.
Należy podać warunki eksperymentu: badane materiały, powierzchnię próbek, skład i stężenie elektrolitów, temperaturę itp.
Wyniki badań należy zapisać w przygotowanych wcześniej tabelach, których wzory podane są w opisie pracy.
Przy rejestrowaniu różnych wielkości konieczne jest wskazanie ich wymiaru. Wyniki pomiarów z reguły poddawane są dodatkowej obróbce – analitycznej i graficznej (obliczenia wskaźników masy, objętości i głębokości korozji, potencjałów itp.). W takich przypadkach konieczne jest podanie wzoru obliczeniowego i jednego obliczenia w całości, tj. z podstawieniem wartości eksperymentalnych do wzoru, a dla innych podobnych obliczeń - tylko wyniki końcowe.
Na podstawie uzyskanych i odpowiednio przetworzonych wyników należy wyciągnąć krótkie wnioski na temat wykonanej pracy. Po zakończeniu prześlij dziennik z danymi eksperymentalnymi nauczycielowi w celu uzyskania wizy.
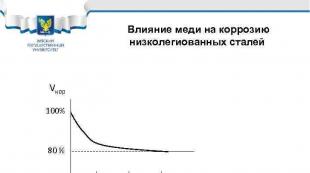
 Wpływ miedzi na korozję stali niskostopowych Vcor 100% 80% 0,1 0,2 0,3% Cu Przykładowe stale: 10 HSND, 10 G 2 S 1 D, 10 KhDNP, 09 G 2 D, 18 G 2 AF(D)
Wpływ miedzi na korozję stali niskostopowych Vcor 100% 80% 0,1 0,2 0,3% Cu Przykładowe stale: 10 HSND, 10 G 2 S 1 D, 10 KhDNP, 09 G 2 D, 18 G 2 AF(D)
 Klasyfikacja stali odpornych na korozję 1. Stale i stopy odporne na korozję (nierdzewne) to materiały odporne na korozję elektrochemiczną w elektrolitach. 2. Głównym pierwiastkiem stopowym odpornym na korozję jest chrom. 3. Chrom wprowadza się do stali nierdzewnych zgodnie z regułą Tammanna. 4. W zależności od środowisk, w jakich stale te są stosowane, wyróżnia się pięć grup stali i stopów odpornych na korozję (nierdzewnych).
Klasyfikacja stali odpornych na korozję 1. Stale i stopy odporne na korozję (nierdzewne) to materiały odporne na korozję elektrochemiczną w elektrolitach. 2. Głównym pierwiastkiem stopowym odpornym na korozję jest chrom. 3. Chrom wprowadza się do stali nierdzewnych zgodnie z regułą Tammanna. 4. W zależności od środowisk, w jakich stale te są stosowane, wyróżnia się pięć grup stali i stopów odpornych na korozję (nierdzewnych).
 Stale odporne na korozję do środowisk średnio agresywnych Stale pierwszej grupy mogą pracować tylko w atmosferze zamkniętej i korozji podwodnej z obowiązkowym okresowym suszeniem. W warunkach otwartej atmosfery i ciągłej korozji podwodnej (zwłaszcza w gorącej wodzie), a także korozji podziemnej stale te ulegają korozji wżerowej. Do tych stali zaliczamy stale chromowe: 08 X 13, 09 X 13, 08 X 17 G (ferrytyczne), 10 X 13, 12 X 13 (martenzytyczno-ferrytyczne), 20 X 13, 30 X 13, 40 X 13 (martenzytyczne). Oraz stale chromowo-manganowe i chromowo-niklowe z ekonomicznym dodatkiem niklu (2 -4%) 15 X 17 AG 14, 10 X 14 AG 15, 10 X 14 G 14 N 3 T, 12 X 17 G 14 N 3 , 08 X 18 G 8 N 2 T
Stale odporne na korozję do środowisk średnio agresywnych Stale pierwszej grupy mogą pracować tylko w atmosferze zamkniętej i korozji podwodnej z obowiązkowym okresowym suszeniem. W warunkach otwartej atmosfery i ciągłej korozji podwodnej (zwłaszcza w gorącej wodzie), a także korozji podziemnej stale te ulegają korozji wżerowej. Do tych stali zaliczamy stale chromowe: 08 X 13, 09 X 13, 08 X 17 G (ferrytyczne), 10 X 13, 12 X 13 (martenzytyczno-ferrytyczne), 20 X 13, 30 X 13, 40 X 13 (martenzytyczne). Oraz stale chromowo-manganowe i chromowo-niklowe z ekonomicznym dodatkiem niklu (2 -4%) 15 X 17 AG 14, 10 X 14 AG 15, 10 X 14 G 14 N 3 T, 12 X 17 G 14 N 3 , 08 X 18 G 8 N 2 T
 Stale odporne na korozję (nierdzewne) do środowisk zasolonych Druga grupa stali odpornych na korozję (nierdzewnych) stosowana jest w środowiskach zasolonych, w niskich temperaturach, szczególnie podczas korozji morskiej. Zwiększoną odporność na korozję uzyskuje się poprzez dodatkowe ekonomiczne domieszkowanie stali Ni (5 – 8%). Przykłady: 09 X 15 N 8 Yu, 07 X 16 N 6, 08 X 17 N 5 M 3 (stal stosowana w środowisku kwasu siarkowego), 09 X 17 N 7 Yu 1 (stal stosowana w warunkach korozji morskiej).
Stale odporne na korozję (nierdzewne) do środowisk zasolonych Druga grupa stali odpornych na korozję (nierdzewnych) stosowana jest w środowiskach zasolonych, w niskich temperaturach, szczególnie podczas korozji morskiej. Zwiększoną odporność na korozję uzyskuje się poprzez dodatkowe ekonomiczne domieszkowanie stali Ni (5 – 8%). Przykłady: 09 X 15 N 8 Yu, 07 X 16 N 6, 08 X 17 N 5 M 3 (stal stosowana w środowisku kwasu siarkowego), 09 X 17 N 7 Yu 1 (stal stosowana w warunkach korozji morskiej).
 Stale do stosowania w środowiskach o średniej korozyjności. Środowiska o średniej korozyjności to roztwory soli o różnych temperaturach, a także słabe roztwory niektórych kwasów. Stale trzeciej grupy są najpowszechniejszymi stalami nierdzewnymi o szerokim zastosowaniu. Wśród tych stali możemy wyróżnić: a) stale - zamienniki stali wysokoniklowych: 15 X 25 T, 15 X 28, 08 X 22 N 6 T, 12 X 21 N 5 T. b) stale o optymalnej zawartości chromu do niklu stosunek (Cr:Ni = 18:9, 18:10): 12 X 18 N 9 T i 12 X 18 N 10 T, 17 X 18 N 9, 12 X 18 N 10 B, 08 X 18 N 10, 12 X 18 N 12 T, 08 X 18 N 12 B, 06 X 18 N 11 itd.
Stale do stosowania w środowiskach o średniej korozyjności. Środowiska o średniej korozyjności to roztwory soli o różnych temperaturach, a także słabe roztwory niektórych kwasów. Stale trzeciej grupy są najpowszechniejszymi stalami nierdzewnymi o szerokim zastosowaniu. Wśród tych stali możemy wyróżnić: a) stale - zamienniki stali wysokoniklowych: 15 X 25 T, 15 X 28, 08 X 22 N 6 T, 12 X 21 N 5 T. b) stale o optymalnej zawartości chromu do niklu stosunek (Cr:Ni = 18:9, 18:10): 12 X 18 N 9 T i 12 X 18 N 10 T, 17 X 18 N 9, 12 X 18 N 10 B, 08 X 18 N 10, 12 X 18 N 12 T, 08 X 18 N 12 B, 06 X 18 N 11 itd.
 Stale do stosowania w środowiskach o zwiększonej korozyjności Te rodzaje stali zostały opracowane w celu zwiększenia odporności chemicznej w gorących roztworach Na. Cl i w roztworach kwasowych. W celu zwiększenia wytrzymałości stali stosuje się dodatkowe domieszki stopowe molibdenem i miedzią, przy czym w stalach tej grupy często dąży się do utrzymania dogodnej technologicznie struktury austenitycznej, która wymaga dodatkowego stopowania stali niklem. Ze względu na dużą zawartość składników stopowych, przede wszystkim niklu, stale tej grupy są dość drogie. Przykładowe stale w tej grupie to: 10 X 17 N 13 M 2 T 08 X 17 N 13 M 3 T, 08 X 17 N 15 M 3 T, 04 X 28 MDT, 03 X 28 MDT, 06 X 28 MT.
Stale do stosowania w środowiskach o zwiększonej korozyjności Te rodzaje stali zostały opracowane w celu zwiększenia odporności chemicznej w gorących roztworach Na. Cl i w roztworach kwasowych. W celu zwiększenia wytrzymałości stali stosuje się dodatkowe domieszki stopowe molibdenem i miedzią, przy czym w stalach tej grupy często dąży się do utrzymania dogodnej technologicznie struktury austenitycznej, która wymaga dodatkowego stopowania stali niklem. Ze względu na dużą zawartość składników stopowych, przede wszystkim niklu, stale tej grupy są dość drogie. Przykładowe stale w tej grupie to: 10 X 17 N 13 M 2 T 08 X 17 N 13 M 3 T, 08 X 17 N 15 M 3 T, 04 X 28 MDT, 03 X 28 MDT, 06 X 28 MT.
 Stopy na bazie niklu do środowisk bardzo agresywnych Przez media o bardzo dużej agresywności rozumie się gorące roztwory kwasów siarkowego i solnego. W tak agresywnym środowisku najbardziej odpornymi materiałami metalowymi są stopy na bazie niklu. Na przykład stop KhN 65 MV jest stabilny w podwyższonych temperaturach w środowisku kwasu siarkowego i solnego, w stężonym kwasie octowym. Stop N 70 MF jest zalecany do stosowania w roztworach kwasu siarkowego i solnego; stop jest bardziej odporny na korozję międzykrystaliczną.
Stopy na bazie niklu do środowisk bardzo agresywnych Przez media o bardzo dużej agresywności rozumie się gorące roztwory kwasów siarkowego i solnego. W tak agresywnym środowisku najbardziej odpornymi materiałami metalowymi są stopy na bazie niklu. Na przykład stop KhN 65 MV jest stabilny w podwyższonych temperaturach w środowisku kwasu siarkowego i solnego, w stężonym kwasie octowym. Stop N 70 MF jest zalecany do stosowania w roztworach kwasu siarkowego i solnego; stop jest bardziej odporny na korozję międzykrystaliczną.
 Zwiększanie gęstości betonu 4. Wprowadzenie dodatków polimerowych 4. 1. wprowadzenie do mieszanki betonowej niewielkiej ilości 0,2 - 3% dodatków polimerowych (lateksy, żywice polimerowe); 4. 2. produkcja betonów na bazie spoiwa polimerowego (roztwory polimerowe i polimerobeton); Dostarczany w postaci suchej mieszanki i utwardzacza w puszkach. 4. 3. impregnacja gotowych wyrobów betonowych i żelbetowych związkami polimerowymi lub monomerami z późniejszą ich polimeryzacją bezpośrednio w korpusie betonowym (polimery betonowe); 4. 4. zbrojenie betonu włóknami polimerowymi (produkcja betonu zbrojonego włóknami)
Zwiększanie gęstości betonu 4. Wprowadzenie dodatków polimerowych 4. 1. wprowadzenie do mieszanki betonowej niewielkiej ilości 0,2 - 3% dodatków polimerowych (lateksy, żywice polimerowe); 4. 2. produkcja betonów na bazie spoiwa polimerowego (roztwory polimerowe i polimerobeton); Dostarczany w postaci suchej mieszanki i utwardzacza w puszkach. 4. 3. impregnacja gotowych wyrobów betonowych i żelbetowych związkami polimerowymi lub monomerami z późniejszą ich polimeryzacją bezpośrednio w korpusie betonowym (polimery betonowe); 4. 4. zbrojenie betonu włóknami polimerowymi (produkcja betonu zbrojonego włóknami)

 Moduł 7. Metody ochrony metali przed korozją elektrochemiczną. Wykład 7.3 ELEKTROCHEMICZNA OCHRONA METALI PRZED KOROZJĄ Ochrona katodowa polega na przesunięciu potencjału metalu korodującej konstrukcji na stronę ujemną poprzez podłączenie go do bieguna ujemnego źródła prądu.
Moduł 7. Metody ochrony metali przed korozją elektrochemiczną. Wykład 7.3 ELEKTROCHEMICZNA OCHRONA METALI PRZED KOROZJĄ Ochrona katodowa polega na przesunięciu potencjału metalu korodującej konstrukcji na stronę ujemną poprzez podłączenie go do bieguna ujemnego źródła prądu.
 Moduł 7. Metody ochrony metali przed korozją elektrochemiczną. Wykład 7.3 Wykres korozji ochrony katodowej
Moduł 7. Metody ochrony metali przed korozją elektrochemiczną. Wykład 7.3 Wykres korozji ochrony katodowej
 Moduł 7. Metody ochrony metali przed korozją elektrochemiczną. Wykład 7.2 Ochrona ochronna opiera się na charakterystyce korozji dwóch stykających się metali. Zgodnie z teorią korozji kontaktowej, gdy dodatni metal M 2 styka się z bardziej ujemnym metalem M 1, potencjał metalu M 2 przesuwa się na stronę ujemną, a jego korozja zmniejsza się lub całkowicie zatrzymuje.
Moduł 7. Metody ochrony metali przed korozją elektrochemiczną. Wykład 7.2 Ochrona ochronna opiera się na charakterystyce korozji dwóch stykających się metali. Zgodnie z teorią korozji kontaktowej, gdy dodatni metal M 2 styka się z bardziej ujemnym metalem M 1, potencjał metalu M 2 przesuwa się na stronę ujemną, a jego korozja zmniejsza się lub całkowicie zatrzymuje.
 Moduł 7. Metody ochrony metali przed korozją elektrochemiczną. Wykład 7.3 Ochronę anodową stosuje się wyłącznie w przypadku metali podatnych na pasywację w środowisku korozyjnym. Polega to na przesunięciu potencjału metalu z obszaru aktywnego rozpuszczania do obszaru pasywacji przy wykorzystaniu zewnętrznego źródła prądu.
Moduł 7. Metody ochrony metali przed korozją elektrochemiczną. Wykład 7.3 Ochronę anodową stosuje się wyłącznie w przypadku metali podatnych na pasywację w środowisku korozyjnym. Polega to na przesunięciu potencjału metalu z obszaru aktywnego rozpuszczania do obszaru pasywacji przy wykorzystaniu zewnętrznego źródła prądu.
 Moduł 7. Metody ochrony metali przed korozją elektrochemiczną. Wykład 7.3 Wykres korozji zabezpieczenia anodowego
Moduł 7. Metody ochrony metali przed korozją elektrochemiczną. Wykład 7.3 Wykres korozji zabezpieczenia anodowego
MINISTERSTWO EDUKACJI I NAUKI FEDERACJI ROSYJSKIEJ PAŃSTWA FEDERALNEGO INSTYTUCJA EDUKACYJNA WYŻSZEJ SZKOLNICTWA ZAWODOWEGO „PAŃSTWOWY UNIWERSYTET TECHNOLOGICZNY POLIMERÓW ROŚLINNYCH W ST. PETERSBURG” T. L. Lukanina, I. Mikhailova, M. A. Radin ODPORNOŚĆ CHEMICZNA MATERIAŁÓW I OCHRONA PRZED KOROZJĄ Podręcznik Sankt-Petersburg 2014 1 UDC 546(075) BBK 24.1я7 L 840 Lukanina T. L., Mikhailova I. S., Radin M. A. Odporność chemiczna materiałów i ochrona przed korozją: podręcznik. podręcznik – St.Petersburg: SPbGTURP, 2014. – 85 s. Podręcznik dostarcza współczesnych pomysłów na temat termodynamiki i kinetyki korozji chemicznej, mechanizmów i różnych rodzajów korozji elektrochemicznej, z uwzględnieniem środowisk i warunków charakterystycznych dla urządzeń przemysłu chemicznego. Przedstawiono naukowe zasady tworzenia stali i stopów odpornych na korozję oraz metody zabezpieczania materiałów konstrukcyjnych przed korozją. Podręcznik przeznaczony jest dla studentów uczelni technicznych i kierunków kształcących się na specjalnościach technologicznych. Może być przydatny dla doktorantów, pracowników naukowych i inżynieryjnych przedsiębiorstw chemicznych, organizacji projektowych i badawczych zajmujących się ochroną metali przed korozją. Recenzenci: Dr Chem. nauki, prof. Katedra Chemii Państwowego ITMO w Petersburgu. wydział Slobodov A. A.; Doktorat chemia nauk ścisłych, profesor nadzwyczajny Katedra Chemii Organicznej, Państwowy Uniwersytet Techniczny w Petersburgu w Turkmenistanie, Evdokimov A.N. Zalecana do publikacji przez Radę Redakcyjną i Wydawniczą Uniwersytetu jako pomoc dydaktyczna. Redaktor i korektor T. A. Smirnova Tech. Redaktor L.Ya. Titova Templan 2014, poz. 45 ____________________________________________________________________________ Do publikacji przesłano 14.04.14 Format 60×84/16. Rodzaj papieru nr 1. Druk offsetowy. Drukowane l.5.5. Uch.-ed. arkusze 5.5. Nakład 100 egz. Wydanie nr 45. Cena „C”. Zamówienie Risograf Państwowego Uniwersytetu Technologicznego Polimerów Roślinnych w Petersburgu, 198095, ul. Ivana Chernykh, 4 St. Petersburg Państwowy Uniwersytet Technologiczny Polimerów Roślinnych, 2014 Lukanina T. L., Mikhailova I. S., Radin M. A., 2014 2 WPROWADZENIE KOROZJA materiałów to samoistne niszczenie na skutek chemicznego lub elektrochemicznego oddziaływania z otoczeniem. Zniszczenie metali i stopów w wyniku korozji różni się od innych rodzajów samorzutnego zniszczenia, takich jak erozja lub zużycie, które są spowodowane mechaniczną interakcją z ciałem lub środowiskiem. W przypadku większości metali i stopów w warunkach pracy stan utleniony (jonowy) jest bardziej stabilny, do którego przechodzą w wyniku korozji. Termin „korozja” pochodzi od łacińskiego słowa „corrodere”, co oznacza „korodować”. Straty powstałe w wyniku korozji dzieli się zazwyczaj na bezpośrednie i pośrednie. STRATY BEZPOŚREDNIE obejmują: - koszt metalu zamieniającego się w produkty korozji, a także koszt skorodowanego sprzętu, który ulegnie awarii przed upływem okresu amortyzacji przewidzianego warunkami technicznymi. Co więcej, z reguły koszt produkcji sprzętu znacznie przewyższa koszt metalu, z którego jest wykonany; − wzrost naddatków na korozję, co prowadzi nie tylko do wzrostu zużycia metalu, ale także do pogorszenia konstrukcji. Na przykład w przypadku rurociągu o tej samej średnicy zewnętrznej zwiększony naddatek metalu prowadzi do zmniejszenia użytecznego przekroju, a w konsekwencji do zmniejszenia przepustowości rury; − koszty środków ochronnych w postaci powłok metalicznych i niemetalicznych, materiałów odpornych na korozję, inhibitorów itp. STRATY POŚREDNIE na skutek korozji powstają z następujących przyczyn: − dodatkowe koszty z tytułu nieplanowanych napraw uszkodzonego sprzętu; − niedobory produkcyjne na skutek przestojów na naprawy urządzeń uszkodzonych korozją; − możliwość wypadków, wybuchów i pożarów; − wycieki produktów na skutek powstawania otworów przelotowych w ściankach urządzeń w wyniku korozji; 3 – zmniejszenie przekroju rurociągów na skutek osadzania się produktów korozji, co wymaga dodatkowego zużycia energii w celu utrzymania wymaganej mocy przepływu płynu; − obniżenie jakości wyrobów na skutek zanieczyszczenia produktami korozji. Nauka o korozji i ochronie metali bada interakcję metali i stopów ze środowiskiem korozyjnym, ustala mechanizm tej interakcji i jej ogólne zasady. Podstawą naukową badań korozji i ochrony metali jest metaloznawstwo i chemia fizyczna. Ostatecznym praktycznym celem tej nauki jest ochrona przed korozyjnym niszczeniem metali podczas ich obróbki i eksploatacji urządzeń. Rozdział 1. OGÓLNE INFORMACJE O KOROZJI 1.1. Termodynamika i kinetyka procesów korozyjnych Podstawową przyczyną korozji jest niestabilność termodynamiczna metali w różnych środowiskach w danych warunkach zewnętrznych. Podstawową możliwość samoistnego wystąpienia procesu korozji wyznaczają prawa termodynamiki. Wystąpieniu dowolnego procesu spontanicznego towarzyszy spadek energii swobodnej. Możliwość samoistnego wystąpienia procesów korozyjnych można ocenić na podstawie zmian potencjału izobaryczno-izotermicznego ΔG1. Spontaniczny proces utleniania metalu - korozji - jest możliwy, jeśli ∆G< 0, то есть знак ―минус‖ означает, что изобарно−изотермический потенциал убывает и свободная энергия системы уменьшается. Наоборот, если ∆G > 0, wówczas znak plus potencjału izobaryczno-izotermicznego odpowiada wzrostowi energii swobodnej, co wskazuje na niemożność spontanicznego wystąpienia tej reakcji. POTENCJAŁ IZOBARYCZNO-izotermiczny (ΔG) – energia Gibbsa, jeden z potencjałów termodynamicznych, równy ΔG = ΔН - TΔS, gdzie ΔН, ΔS, Т - entalpia, entropia, temperatura termodynamiczna układu. ΔG jest proporcjonalne do liczby cząstek w układzie. Określany jako jedna cząstka, nazywany jest potencjałem chemicznym. ΔG jest wygodny do opisu procesów, w których możliwa jest wymiana między materią a otaczającymi ją ciałami. W procesie równowagi izotermicznej zachodzącym pod stałym ciśnieniem jest to całkowita praca układu pomniejszona o pracę przeciw zewnętrznym siłom ciśnienia. Jest równy spadkowi potencjału izobaryczno-izotermicznego (tj. maksymalnej użytecznej pracy). 1 4 Jednakże, choć termodynamika pozwala określić, jak daleko badany układ znajduje się od stanu równowagi i jest zdolny do spontanicznej reakcji, nie oznacza to szybkości tego procesu. Szybkość korozji zależy od jej kinetyki. Cechą procesów korozyjnych jest ich niejednorodność. Wynika to z faktu, że niszczenie metalu następuje na granicy faz pomiędzy dwiema fazami, które mają różne stany skupienia, np. na granicy faz metal-ciecz, metal-gaz. W takich warunkach czynnikiem determinującym kinetykę oddziaływania chemicznego jest albo szybkość oddziaływania chemicznego, albo dopływ odczynników z objętości stykających się faz do powierzchni międzyfazowej, na której zachodzi oddziaływanie korozyjne, oraz usuwanie produktów reakcji w kierunku przeciwnym kierunek. W pierwszym przypadku szybkość korozji określa się poprzez kontrolę kinetyczną, w drugim - poprzez kontrolę dyfuzyjną. W niektórych przypadkach szybkość korozji określa się na podstawie mieszanej kontroli dyfuzyjno-kinetycznej. W przypadku procesu w stanie ustalonym szybkość reakcji chemicznej i szybkość dyfuzji są równe. Proces korozji składa się z kilku etapów, które mogą zachodzić sekwencyjnie lub równolegle. Całkowita szybkość procesu korozji składającego się z kilku kolejnych etapów w stanie ustalonym jest określona przez szybkość najwolniejszego etapu. Gdy poszczególne etapy procesu zachodzą równolegle, o całkowitej szybkości korozji decyduje etap najszybszy. Dlatego jeśli jeden z etapów przebiega znacznie szybciej niż drugi, drugi etap można pominąć. 1.2. Klasyfikacja procesów korozyjnych ze względu na mechanizm ich występowania Niezależnie od ogromnej różnorodności warunków występowania procesów korozyjnych, można je klasyfikować według dwóch głównych cech: - ze względu na mechanizm procesu - ze względu na charakter zniszczenia korozyjnego . Wyróżnia się korozję CHEMICZNĄ i ELEKTROCHEMICZNĄ. 5 występuje w suchych gazach (korozja gazowa) i w ciekłych nieelektrolitach2 (korozja w nieelektrolitach). Jeśli ośrodkiem nie jest elektrolit (suche gazy lub ciekłe nieelektrolity), wówczas wymiana elektronów zachodzi bezpośrednio pomiędzy metalem redukującym (czerwonym) a dowolną substancją zawierającą pierwiastek elektroujemny zdolny do przyciągania do siebie elektronów - utleniacza (wół) ), na przykład: Fe + Сl2 → FeCl2 (1) KOROZJA CHEMICZNA czerwony Bilans przeniesienia elektronów wół Fe° − 2ē → Fe2+ 1 Сl2° + 2ē → 2Cl− Fe° + 2Сl° = Fe2+ + 2Cl− KOROZJA ELEKTROCHEMICZNA to proces oddziaływania metalu z ośrodkiem korozyjnym (roztworem elektrolitu), w którym jonizacja atomów metalu i redukcja składnika utleniającego środowiska korozyjnego nie zachodzą w jednym procesie, a ich szybkości zależą od potencjału elektrody. Cechami charakterystycznymi procesu korozji elektrochemicznej są: – jednoczesne występowanie dwóch odrębnych procesów – utleniającego (rozpuszczanie metalu) i redukcyjnego (wydzielanie się wodoru, redukcja tlenu, uwalnianie metalu z roztworu itp.); – procesowi rozpuszczania metalu towarzyszy kierunkowy ruch elektronów w metalu i jonów w elektrolicie, tj. występowanie prądu elektrycznego; – produkty korozji powstają w wyniku reakcji wtórnych. Procesy redoks zachodzące podczas korozji elektrochemicznej można przedstawić w postaci następujących reakcji: Ме − nē → Меn+ (proces anodowy) (2) n− R(ox) + nē → R (czerwony) (proces katodowy), (3 ) gdzie R(ox) oznacza środek utleniający; NIEELEKTROLITY to substancje, których wodne roztwory i stopy nie przewodzą prądu elektrycznego. Zawierają kowalencyjne wiązania niepolarne lub niskopolarne, które nie rozkładają się na jony. Gazy, ciała stałe (niemetale) i związki organiczne (sacharoza, benzyna, alkohol) nie przewodzą prądu elektrycznego. 2 6 R n−(red) – zredukowana forma utleniacza; nē – liczba przeniesionych elektronów. Przykładem korozji elektrochemicznej jest proces utleniania (rdzewienia) żelaza pod wpływem wody: Fe − 2ē → Fe2+ (proces anodowy - rozpuszczanie żelaza) H2O + ½О2 +2ē → 2OH– (proces katodowy - redukcja tlenu) −−−−−− −−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−− − Fe 2+ + 2OH – → Fe(OH)2 (powstanie produktów korozji) Reakcje (2) i (3) przebiegają w sposób sprzężony, ale podlegają własnym prawom kinetycznym. W takim przypadku konieczne jest zachowanie warunków stacjonarności procesu, tj. równość szybkości utleniania metalu i redukcji środka utleniającego. Reakcje te można rozdzielić geograficznie i zachodzić w różnych częściach powierzchni. Z warunków stacjonarności wynika, że wystarczy spowolnić jedną z reakcji sprzężonych, aby szybkość całego procesu spadła. 1.3. Klasyfikacja procesów korozyjnych ze względu na charakter niszczenia korozyjnego W oparciu o charakter niszczenia korozyjnego metalu wyróżnia się KOROZJĘ OGÓLNĄ I MIEJSCOWĄ. KOROZJA OGÓLNA rozprzestrzenia się na całej powierzchni metalu, może być jednolita lub nierówna (ryc. 1. - 1.2). 1 2 4 5 7 8 3 6 9 Rys. 1. Rodzaje uszkodzeń korozyjnych Wiele przypadków korozji obejmuje korozję ogólną, np. gdy w wyniku wystawienia metalu na działanie elektrolitów produkty korozji nie pozostają na powierzchni metalu. Zatem intensywną korozję ogólną obserwuje się podczas oddziaływania miedzi z kwasem azotowym, żelaza z kwasem solnym, aluminium z zasadami żrącymi, cynku z kwasem siarkowym itp. Korozja ogólna równomierna jest jednym z najmniej niebezpiecznych rodzajów korozji, pod warunkiem, że szybkość rozpuszczania metalu w wyniku uszkodzeń korozyjnych nie przekracza norm określonych w wymaganiach dotyczących działania aparatu, urządzenia itp. Jeżeli metal jest wystarczająco gruby, korozja ma niewielki wpływ na zmniejszenie wytrzymałości mechanicznej konstrukcji pod wpływem równomiernie rozłożonych naprężeń w przekroju poprzecznym (rozciąganie, ściskanie). Jednak ogólna korozja może być niebezpieczna, gdy części są poddawane zginaniu i skręcaniu, ponieważ w tym przypadku najbardziej obciążone warstwy ulegają zniszczeniu. KOROZJA MIEJSCOWA - powoduje zniszczenie tylko niektórych obszarów powierzchni metalu, a reszta nie ulega zniszczeniu lub stopień zniszczenia jest bardzo niski. Objawy korozji miejscowej są bardzo zróżnicowane i mogą mieć różny stopień nierówności, dlatego też korozja lokalna dzieli się na KOROZJĘ PRZEZ PLAMA, WRODY, NACZYNIA (PITTING), KOROZJĘ SELEKTYWNĄ, KOROZJĘ, PĘKANIE KOROZYJNE. MIĘDZYKRYSTALITOWA Korozja punktowa, wrzodowa, punktowa – rodzaje korozji miejscowej różnią się stosunkiem średnicy zniszczonego obszaru do jego głębokości (ryc. 1, 4–6). W przypadku korozji punktowej średnica zniszczonego obszaru jest znacznie większa niż jego głębokość, w przypadku wrzodziejącego zniszczenia średnica zniszczonego obszaru jest proporcjonalna do jego głębokości, a w przypadku korozji wżerowej średnica zniszczonego obszaru jest znacznie mniejsza niż jego głębokość . W niektórych przypadkach korozja wżerowa może skutkować zniszczeniem, co jest szczególnie niebezpieczne dla rurociągów, zbiorników itp. Korozja wżerowa jest typowa dla metali zdolnych do pasywacji - hamowania (chrom, aluminium, stal nierdzewna itp.). Stan pasywny metali może zostać zakłócony w pewnych słabszych obszarach powierzchni, np. w porach warstwy ochronnej, w miejscach wtrąceń międzymetalicznych (tlenki, żużle itp.). KOROZJĘ SELEKTYWNĄ – dzieli się na: selektywną składową (selektywną) lub selektywną strukturalną (międzykrystaliczną). 8 Przykładem KOROZJI SELEKTYWNEJ KOMPONENTÓW jest odcynkowanie mosiądzu. Odcynkowanie mosiądzu3 polega na tym, że cynk przenika do elektrolitu (zwykle przy obojętnym lub lekko kwaśnym pH) intensywniej niż miedź. Na powierzchni mosiądzu tworzy się luźna warstwa miedzi, która przyczynia się do wzmożonej korozji elektrochemicznej (pojawienie się ogniwa mikrogalwanicznego). Przykładem korozji KONSTRUKCYJNEJ SELEKTYWNEJ jest korozja żeliwa szarego. Korozja selektywna strukturalna żeliwa szarego polega na przeważającym zniszczeniu składnika ferrytowego, w wyniku czego powstaje szkielet grafitowy wypełniony produktami korozji (rys. 1-3). Jednocześnie wytrzymałość mechaniczna żeliwa gwałtownie maleje. KOROZJA MIĘDZYKRYSTALITOWA charakteryzuje się selektywnym niszczeniem metalu wzdłuż granic ziaren (rys. 1–7). W rezultacie przy niewielkiej zmianie masy i wyglądu może nastąpić bardzo znacząca utrata właściwości wytrzymałościowych metalu (ryc. 2). 40 Ryc. 2. Utrata właściwości wytrzymałościowych metalu podczas różnych rodzajów zniszczenia korozyjnego 1 – korozja równomierna; 2 – korozja międzykrystaliczna 20 Utrata w, % 60 2 0 1 2 4 Ubytek masy, kg/cm2 W skrajnych przypadkach, przy całkowitym zniszczeniu granic ziaren w wyniku korozji międzykrystalicznej, stop może rozpaść się na proszek. PĘKANIE KOROZJOWE to przypadek korozji miejscowej, w której zniszczenie następuje w kierunku prostopadłym do największego naprężenia rozciągającego (rys. 1 - 8). 3 Mosiądz – stop miedzi i cynku. 9 Typowe jest, że w tym przypadku pęknięcie korozyjne może rozprzestrzeniać się wzdłuż granic ziaren lub przecinać ciało kryształu. Zniszczenie spowodowane zmęczeniem korozyjnym (przy jednoczesnym narażeniu na środowisko korozyjne i obciążenia zmienne lub cykliczne) przebiega w podobny sposób, jak podczas pękania korozyjnego. KOROZJA PODPOWIERZCHNIOWA, rozpoczynając od powierzchni, rozprzestrzenia się głównie pod powierzchnią, co często prowadzi do rozwarstwiania i pęcznienia metalu (rys. 1-9), np. tworzenia się pęcherzyków na niskiej jakości blachach w procesie trawienia lub korozja podczas eksploatacji zbiorników i aparatury. 1.4. Wskaźniki korozji Do ilościowego określenia szybkości korozji stosuje się wskaźniki korozji: MASOMETRYCZNE, OBJĘTOŚCIOWE, GŁĘBOKOŚCIOWE, MECHANICZNE, PRĄDOWE. 2 MASSOMETRYCZNY wskaźnik korozji (Km, g/m ∙h) to zmiana masy metalu w wyniku korozji na jednostkę powierzchni próbki i na jednostkę czasu. Zmianę masy próbki określa się zazwyczaj jako różnicę pomiędzy masą próbki przed badaniem (mо) a jej masą po badaniu (m1) w czasie (τ) z usunięciem produktów korozji (utrata masy metalu) . K. m = (m0 - m1)/s ∙ τ . (4) 3 2 OBJĘTOŚCIOWY wskaźnik korozji (Kν, cm /m h) to objętość (Vo) gazu pochłoniętego lub uwolnionego w procesie korozji, na jednostkę powierzchni metalu i na jednostkę czasu K m = V0 /s ∙ τ. (5) Wskaźnik korozji GŁĘBOKOŚCI (P, mm/rok) uwzględnia zmniejszenie grubości metalu w wyniku korozji, wyrażone w jednostkach liniowych i na jednostkę czasu. Wskaźnik ten jest przydatny przy porównywaniu korozji metali o różnych gęstościach. Przejścia od wskaźnika masometrycznego do głębokości można dokonać w przypadku korozji równomiernej korzystając ze wzoru P = K m ∙ 8,76 /γ, (6) gdzie γ jest gęstością metalu. Wskaźnik korozji MECHANICZNEJ określa się na podstawie zmiany jednego z głównych wskaźników właściwości mechanicznych metalu w określonym czasie procesu korozji i wyraża się go w jednostkach względnych lub procentowo. Jeżeli jako wskaźnik mechaniczny przyjmuje się wytrzymałość na rozciąganie, wówczas zmianę wskaźnika wytrzymałości (Kσ) określa się wzorem Kσ = Δσb/σb, (7) gdzie σb to wytrzymałość na rozciąganie przed korozją, ∆σb to zmiana wytrzymałości na rozciąganie metalu po korozji w czasie τ. Aktualny wskaźnik korozji pozwala, korzystając ze wzoru Faradaya, obliczyć ilość skorodowanego metalu na podstawie wielkości prądu korozyjnego Km = IA /nF, (8) gdzie Km jest szybkością korozji na podstawie zmiany w masie metalicznej; I – aktualna siła; F – stała Faradaya; n – wartościowość metalu; A jest masą atomową metalu. Przy jakościowej i ilościowej ocenie odporności metali na korozję zaleca się stosowanie skali dziesięciopunktowej4. Należy zauważyć, że chociaż ta skala oceny odporności metali na korozję stała się bardziej rozpowszechniona, zwłaszcza w przemyśle chemicznym, nie jest ona uniwersalna, ponieważ w przypadku sprzętu do różnych celów dopuszczalne szybkości korozji mogą różnić się o rząd wielkości lub więcej. Tabela 1 Dziesięć Skala kulowa odporności na korozję metali i stopów Grupa odporności całkowicie trwała, bardzo trwała odporna niska odporność niestabilna prędkość korozji, mm/rok mniej niż 0,001 ponad 0,001 do 0,005 do 0,01 do 0,01 do 0,05 ponad 0,05 więcej 05 do 0,1 ponad 0,1 do 0,5 ponad 0,5 do 1,0 ponad 1,0 do 5,0 ponad 5,0 do 10,0 ponad 10,0 OCENA 1 2 3 4 5 6 7 8 9 10 Ujednolicony system ochrony przed korozją i starzeniem. Konstrukcje podziemne. Ogólne wymagania dotyczące ochrony przed korozją. GOST 9.602−2005, http://www.gosthelp.ru/gost/gost206.html 4 11 Rozdział 2. KOROZJA CHEMICZNA METALI Korozja chemiczna metali jest samoistnym procesem ich niszczenia w wyniku interakcji chemicznej ze środowiskiem zewnętrznym. Cechami charakterystycznymi procesu korozji chemicznej są: – realizacja procesów utleniania i redukcji w jednym etapie; – brak prądu elektrycznego; – powstawanie produktów korozji bezpośrednio na powierzchni, gdzie następuje jej zniszczenie. KOROZJA CHEMICZNA to niejednorodna reakcja oddziaływania fazy ciekłej (l) lub gazowej (g) z fazą stałą (fazą) - metalem i dzieli się na 2 typy (korozja gazowa, korozja w ciekłych nieelektrolitach). 2.1. Korozja gazowa W praktyce największe znaczenie ma KOROZJA GAZOWA. Najczęściej objawia się korozją materiałów metalowych w wysokich temperaturach (kilkadziesiąt razy wyższych od temperatury wrzenia wody) w atmosferze gazów zawierających tlen. Z tego powodu nazywa się to KOROZJĄ W WYSOKIEJ TEMPERATURZE lub UTLENIANIEM W WYSOKIEJ TEMPERATURZE. Na powierzchni wyrobów wykonanych z metali i stopów, w wyniku ich interakcji z tak korozyjnym środowiskiem, tworzy się film produktów reprezentujących różne tlenki metali. Jednocześnie, w zależności od warunków pracy, mogą powstawać inne związki, na przykład metale siarkowe5. Na przykład w przemyśle rafinacji ropy naftowej korozja gazowa wpływa na przykład na rury pieców, ściany reaktora, kolumny reakcyjne i inny sprzęt narażony na działanie siarkowodoru, tlenku węgla, azotu, tlenu i innych gazów w wysokich temperaturach. Termin „korozja gazowa” odnosi się do niszczenia metali spowodowanego działaniem par i gazów w wysokich temperaturach, chociaż można go również stosować w niższych temperaturach przy 5 SIARKACH METALI, takich samych jak siarczki. 12 należy upewnić się, że na metalu nie skrapla się warstwa cieczy przewodzącej prąd elektryczny. Na powierzchni wyrobów wykonanych z metali i stopów, w wyniku ich interakcji ze środowiskiem korozyjnym, tworzy się film produktów reprezentujących różne związki metali. Ogólnie proces korozji chemicznej można opisać np. reakcją utleniania metalu z podgrzanym (T >>373K) gazem (G2): 2Ме + nГ2 → 2Men+Гn. W środowisku tlenowym proces taki można opisać równaniem Me(T) + ½ O2 ↔ MeO(T). Ta reakcja jest równowagowa w wysokich temperaturach. Proces odwrotny nazywa się ROZWOJEM TLENKU. Tlen obecny w środowisku gazowym charakteryzuje się ciśnieniem cząstkowym (pO2). Reakcja chemiczna utleniania metalu będzie w równowadze z reakcją dysocjacji tlenku, jeśli ciśnienie cząstkowe tlenu (pO2) i ELASTYCZNOŚĆ DSOCJACJI TLENKU (pMeO)7 zrównają się (pMeO = pO2) 1. Gdy pO2 > pMeO, reakcja przebiega głównie w kierunku tworzenia tlenków. Gdy pMeO > pO2, reakcja przebiega przeważnie w przeciwnym kierunku. Utlenianie metali jest wieloetapowym, heterogenicznym procesem, w wyniku którego najpierw na powierzchni tworzy się jedno-, a następnie wielocząsteczkowa warstwa tlenku, tzw. film tlenkowy. Wzrost jego grubości może następować albo w obu kierunkach jednocześnie (metal i ośrodek gazowy), albo przeważnie w jednym (ryc. 3). CIŚNIENIE CZĘŚCIOWE (od późn. łac. Partialis - częściowe), ciśnienie, jakie miałby gaz zawarty w mieszaninie gazów, gdyby sam zajmował objętość równą objętości mieszaniny w tej samej temperaturze. 7 Równowagowe ciśnienie cząstkowe tlenu dla reakcji tworzenia lub TERMICZNEJ DYSOCJACJI TLENKÓW nazywa się ELASTYCZNOŚCIĄ DYSOCJACJI TLENKU. Jego wartość jest związana ze stałą równowagi reakcji i dlatego jest funkcją temperatury. 6 13 a b c Rys. Ryc. 3. Schemat jonowo-elektronicznego mechanizmu powstawania i wzrostu warstwy tlenku na powierzchni części metalowej. Jeżeli wzrost następuje w kierunku ośrodka gazowego, obserwuje się znaczny wzrost wielkości części na przykład podczas utleniania, co należy wziąć pod uwagę w inżynierii mechanicznej. Kierunek wzrostu jest określony przez stosunek szybkości przeciwdyfuzji jonów metali Мen+ i tlenu O2− wewnątrz warstwy tlenkowej. Jeśli szybkości dyfuzji jonów metali (rdif.Me) i tlenu (rdif.O2) różnią się znacznie, wówczas wzrost warstwy tlenkowej zachodzi głównie w jednym kierunku, np. przy rdiff.O2< rдиф.Mе − в направлении газовой среды и наоборот (рис. 3, в, б). В большинстве случаев, скорости диффузии ионов металла и кислорода соизмеримы, но для ионов кислорода скорость несколько ниже из−за того, что они больше по размерам, поэтому зона роста, находящаяся в толще самой оксидной пленки, располагается ближе к ее внешней поверхности (рис. 3, а). По мере утолщения оксидной пленки процессы встречной диффузии ионов и электронов затрудняются. С повышением температуры упругость диссоциации оксидов возрастает у всех металлов. Поэтому, начиная от какой−то температуры, когда упругость диссоциации оксида металла становится больше парциального давления кислорода в воздухе (при атмосферном давлении рО2 = 0,21 ат), металл перестает окисляться и становится вполне малоактивным по отношению к кислороду. Так, например, серебро при температуре 300 ºК термодинамически еще не устойчиво, а при 400 ºК и при всех более высоких температурах упругость диссоциации Ag2O превышает парциаль14 ное давление кислорода в воздухе. При температуре 2000 ºК медь тоже становится неокисляемым металлом, однако для таких металлов, как Fе, Zn, Ni и др., даже при этих температурах упругость диссоциации окислов остается еще достаточно низкой и, следовательно, протекание реакции окисления вероятно. Необходимо учесть, что температурная зависимость для кинетики процесса окисления совершенно иная, чем для термодинамической вероятности окисления. Поэтому нет никакого противоречия в том, что термодинамическая вероятность окисления металла с повышением температуры падает, а реальная скорость окисления (при условии, что термодинамически этот процесс остается еще возможным) с повышением температуры возрастает. 2.2. Образование тонких пленок на металлах Ранее было отмечено, что в большинстве случаев образовавшиеся продукты газовой коррозии – оксиды металла остаются на поверхности металла в виде пленки. Эти пленки определяют кинетику процесса и в случае наличия защитных свойств могут привести к замедлению коррозии. Чтобы пленка оксида металла обладала защитными свойствами, она должна быть: − сплошной; − прочно сцепляться с основным металлом и обладать близким к нему коэффициентом теплового расширения; − быть химически инертной по отношению к данной агрессивной среде; − обладать твердостью и износостойкостью. Даже при обычной температуре на поверхности многих металлов и сплавов на воздухе образуются слои оксидов. Еще в 1836 г. М. Фарадей высказал предположение, что даже на блестящей поверхности металлов существует тончайшая, невидимая защитная (оксидная) пленка. Этим он объяснял пассивность железа в азотной кислоте. Если пленка пористая, рыхлая и характеризуется плохим сцеплением с основным металлом, то даже при условии инертности в данной агрессивной среде, она не будет обладать защитными свойствами. Пленки по толщине (δ) делятся на три группы: − тонкие (невидимые), δ < 4000 нм; − средние (дающие цвета побежалости), δ = от 4000 до 50000 нм; 15 − толстые (видимые), δ > 50000 nm. Grubość filmów powstałych podczas oddziaływania metalu z suchym powietrzem jest różna i zależy od rodzaju metalu, temperatury i innych czynników. Zatem grubość folii na miedzi i żelazie w temperaturze pokojowej wynosi 100–300 nm, a na aluminium 1000–1500 nm. Podajmy kilka przykładów potwierdzających obecność cienkich warstw na metalach. 1. Gdy czysta, błyszcząca powierzchnia żelaznej płyty zostanie podgrzana z jednej strony, pojawią się na niej kolory interferencyjne - „kolory matowe”. Wskazuje to na pojawienie się tlenków o różnej grubości. Kolory następują w następującej kolejności – od zimnego końca płytki do gorącego, odpowiednio: żółty, pomarańczowy, czerwony, fioletowy, fioletowy, niebieski. Podczas badania właściwości ochronnych utworzonej folii kroplami roztworu azotanu miedzi, w miejscach, gdzie folia ma słabe właściwości ochronne, reaguje z żelazem: Cu(NO3)2 + Fe → Fe(NO3)2 + Cu. W rezultacie miedź pojawi się na powierzchni pod kroplami. Do czasu, po którym nastąpi reakcja, można ocenić właściwości ochronne folii w różnych obszarach. W tym doświadczeniu najlepsze właściwości ochronne stwierdzono w obszarze oddalonym od miejsca nagrzewania (za żółtą strefą), gdzie powierzchnia pozostaje niezmieniona. Oczywiste jest, że w tym obszarze znajduje się bardzo cienki, ale gęstszy film w porównaniu do obszarów ogrzewanych. 2. Jeśli złamiesz igłę (lub żyletkę) pod wpływem rtęci, w złamanych miejscach utworzy się amalgamat. Po umieszczeniu rozbitych próbek w powietrzu zawierającym rtęć nie obserwuje się amalgamacji. Wyjaśnia to tworzenie się bardzo cienkiej warstwy tlenku, która zapobiega amalgamacji. 3. Po umieszczeniu żelaza w roztworze wodorotlenku potasu z nadmiarem jodu na powierzchni żelaza w roztworze powstaje głębokie zadrapanie. Po pewnym czasie żelazo w miejscu zarysowania zaczyna się rozpuszczać, a na dnie szkła pozostają płatki, które po dalszym badaniu okazują się być tlenkiem żelaza(III). Proces tworzenia filmu na metalu zachodzi etapami. 16 Pierwszym etapem procesu oddziaływania metalu ze środowiskiem korozyjnym jest chemisorpcja środka utleniającego (O2, CO2, H2O, SO2) na powierzchni metalu, np.: Me(sol) + (O2) = Me( sol) + 2 ładunki. W obecności powinowactwa chemicznego pomiędzy metalem a utleniaczem realizowany jest drugi etap oddziaływania metalu ze środowiskiem korozyjnym - przejście warstwy chemisorbowanej do warstwy tlenkowej. Proces ten można umownie opisać reakcją w postaci: xMe(s) + yOads = MexOy. Warstwa tlenkowa utworzona na powierzchni metalu może spowolnić proces korozji na skutek zahamowania dopływu środka utleniającego do powierzchni metalu utleniającego. W tym przypadku folia MA WŁAŚCIWOŚCI OCHRONNE. 2.2.1. Warunek ciągłości folii na metalach Właściwości ochronne mogą mieć jedynie FOLIE CIĄGŁE. Możliwość powstania takiego filmu określa WARUNEK CIĄGŁOŚCI Pillinga-Bedwortha. Objętość molowa związku pojawiająca się na powierzchni metalu (Vok) musi być większa niż objętość metalu (VMe) zużytego do utworzenia jednego mola związku: Vok/VMe >1. Jeśli Vok/VMe< 1, то пленка не может быть сплошной. К металлам, не удовлетворяющим условию сплошности при окислении их кислородом, относятся щелочные и щелочно−земельные металлы. Сплошность пленок – необходимый, но недостаточный фактор, определяющий ее защитные свойства. Как показал Францевич И. Н., у пленок с Vok/VMe >>1 nie mogą mieć wysokich właściwości ochronnych, ze względu na występowanie w nich dużych naprężeń wewnętrznych (np. MoO3 lub WO3). Przyjmuje się, że górna granica stosunku objętościowego wynosi 2,5. Następnie dopracowany warunek ciągłości wygląda następująco: 1< Vok/VMe < 2,5. 2.2.2. Законы роста оксидных пленок на металлах Процесс роста ПОРИСТОЙ ПЛЕНКИ не осложняется процессом подвода окислителя к поверхности металла, а контролируется скоростью химической реакции образования оксида. Скорость коррозии в этом случае выражается уравнением: 17 V = dx/d = kC, где x − толщина пленки; – время; k − константа скорости химической реакции образования оксида; C − концентрация окислителя на поверхности метала. Рост пористой пленки, контролируемый скоростью химической реакции окисления металла, протекает во времени ПО ЛИНЕЙНОМУ ЗАКОНУ. Линейный закон роста имеет место: − при высокотемпературном окислении на воздухе и в среде кислорода при Vok/VMe<1; − в случае образовании летучих оксидов. Сплошные пленки затрудняют проникновение реагентов друг к другу и их рост сопровождается самоторможением процесса. Результирующая скорость этого сложного процесса определяется скоростью самой медленной стадии, т.е. возможны различные варианты контроля течения процесса. 1−й вариант. Скорость коррозии контролируется стадией массопереноса (диффузионный контроль процесса). В этом случае концентрация окислителя на границе раздела Ме – оксидная пленка равна нулю, так как скорость химической реакции весьма велика. Уравнение скорости течения коррозии определяется ПАРАБОЛИЧЕСКИМ ЗАКОНОМ роста оксидной пленки. Параболический закон роста реализуется, в частности, при окислении железа на воздухе при различных температурах. 2−ой вариант. В предыдущем случае С = 0, т.е. накопление окислителя на внутренней поверхности оксидной пленки не происходит. Если учесть возможность кинетического торможения процесса роста оксидной пленки наряду с торможением на стадии массопереноса (диффузионно−кинетический контроль процесса), в этом случае приходим к СЛОЖНО − ПАРАБОЛИЧЕСКОМУ ЗАКОНУ роста оксидной пленки. Рост оксидных пленок по сложно−параболическому закону выполняется: а) при окислении железа в парах воды или в атмосфере СО2; б) при окислении Сo, Сu в атмосфере чистого кислорода; в) при частичном разрушении оксидной пленки. Рост оксидных пленок при диффузионно−кинетическом контроле может быть выражен ТАКЖЕ СТЕПЕННЫМ ЗАКОНОМ. 18 3−й вариант. Часто рост пленки протекает медленнее, чем это следует из параболического закона роста. Это наблюдается при возникновении оксидных пленок при низких температурах (на Сu в О2 при t < 100 оС; на Аl, Тi, Ni, Zn в О2 при t = 300400 оС.) Затухание объясняется либо уплотнением пленок, либо появлением дефектов (пузырей, расслоений). В этих случаях рост толщины пленки протекает в соответствии с логарифмическим законом роста: x = ln(K). 2.3. Термодинамика процесса химической коррозии В качестве примера рассмотрим процесс окисления металла в атмосфере кислорода. Процесс окисления металла может быть представлен реакцией вида: 2Ме + nО2 → Me2n+Оn. Движущая сила процесса коррозии (ДСП) может быть определена с помощью термодинамики. Торможение процесса (ТП) не может быть определено термодинамически, однако с помощью термодинамики можно оценить условия, уменьшающие или исключающие протекание процесса (применение защитных сред, катодная защита, обескислороживание и др.). Независимо от механизма коррозии возможность ее протекания определяется знаком изменения термодинамического потенциала (см. раздел 1.1). Любое изменение энергии системы характеризует переход ее в новое состояние и является мерой стабильности системы: ΔG = GII − GI , где ΔG – энергия, израсходованная на изменение состояния системы, например, на процесс коррозии; GI − энергия системы в исходном состоянии, например, металла в конкретных условиях; G II − энергия системы в новом состоянии, например, прокорродировавшего металла. Рассчитать ΔG можно, вспомнив начало химии8. Величину изобарно-изотермического потенциала можно рассчитать по разности между суммами Δ G продуктов реакции и исходных веществ: GХР = ΔG ni – ΔG mi. (прод. р-ции) (исх. в-в). ΔG - является функцией температуры. Стандартные значения для большинства веществ сведены в справочные таблицы термодинамических величин. Изменение энергии Гиббса в зависимости от температуры учитывается уравнением Гиббса. ΔG Т = ΔН 298 – 298 Δ S 298. ΔG простого вещества = 0. Определить энергию Гиббса для любой химической реакции можно по уравнению ΔG = ΔG + RТ ln К, рассчитав предварительно величину константы равновесия, например, по равновесным концентрациям веществ. При равновесии ΔG=0, поэтому ΔG+RТlnК=0, следова8 19 2.4. Кинетика процесса химической коррозии Хотя термодинамика и дает ответ на вопрос о том, насколько изучаемая система отдалена от состояния равновесия, но ответ на весьма важный практический вопрос о скорости протекания коррозионного разрушения в рамках термодинамики получен быть не может. Рассмотрением этого вопроса и занимается кинетика коррозионных процессов. 2.4.1. Скорость процесса коррозии. Показатели химической коррозии СКОРОСТЬ КОРРОЗИОННОГО процесса может быть представлена в общем виде с помощью уравнения: СКОРОСТЬ КОРРОЗИИ (СК) = ДСП / ТП, где ДСП − движущая сила процесса; ТП − торможение процесса. Скорость химической коррозии определяют количественно по наблюдениям во времени за изменением какой−либо подходящей для этих целей величины, изменяющейся в процессе коррозии. Тогда истинная скорость коррозии металлического материала определяется из выражения: V = dy/d, где y − изменяющаяся в процессе коррозии характеристика свойств материала; − время коррозии. Для количественного выражения скорости коррозионных разрушений на практике пользуются показателями коррозии, являющимися, по сути, отражением СРЕДНЕЙ СКОРОСТИ КОРРОЗИОННОГО разрушения материала: Vср=∆y/∆. Показатели коррозии. 1. ГЛУБИННЫЙ ПОКАЗАТЕЛЬ коррозии: Kn=∆П/∆, где ∆П − глубина (средняя или максимальная) коррозионного разрушения; ∆ − промежуток времени коррозии. 2. ПОКАЗАТЕЛЬ ИЗМЕНЕНИЯ ТОЛЩИНЫ образующейся на металле пленки продуктов коррозии: Kn= ∆h/∆, где ∆h − изменение толщины образующейся на металле пленки продуктов коррозии. тельно, ΔG = – RТlnК. Данное уравнение позволяет также определить значение константы равновесия химической реакции по величине изобарно-изотермического потенциала. Для определения направления протекания многих химических реакций достаточно оценить знак Δ G. 1). При ΔН 0 , Δ S 0 реакция самопроизвольна (Δ G 0) при Т. 2). При ΔН 0, Δ S 0 реакция обратная (Δ G 0) при любой Т. 3). При ΔН 0, Δ S 0, реакция самопроизвольна (Δ G 0) при любой Т. 4). При ΔН 0, ΔS 0 реакция самопроизвольна (Δ G 0) при Т. 20 3. ПОКАЗАТЕЛЬ ИЗМЕНЕНИЯ МАССЫ Km=∆m/S∆, где ∆m − изменение массы корродирующего металла; S – площадь поверхности коррозии. 4.ОБЪЕМНЫЙ ПОКАЗАТЕЛЬ КОРРОЗИИ Kv=∆V/S∆, где ∆V − объем газа, выделившегося или поглощенного в процессе коррозии и приведенный к нормальным условиям. 5. МЕХАНИЧЕСКИЙ ПОКАЗАТЕЛЬ КОРРОЗИИ Кs=(∆s/∆)100 %, где ∆s − относительное изменение характеристики механического свойства. 6. ПОКАЗАТЕЛЬ ИЗМЕНЕНИЯ ЭЛЕКТРИЧЕСКОГО СОПРОТИВЛЕНИЯ KR=(∆R/∆)100 %, где ∆R − относительное изменение электросопротивления образца. 2.4.2. Влияние внешних факторов на химическую коррозию металлов Скорость и характер процесса химической коррозии металлов зависит от ряда факторов. ВНЕШНИМИ ФАКТОРАМИ называют те, которые связаны с составом коррозионной среды и условиями коррозии (температура, давление, скорость перемещения коррозионной среды и т.д.). Температура − мощный внешний фактор коррозии. Характер влияния температуры на скорость окисления металла определяется зависимостью константы скорости реакции окисления (k) и диффузии (D) от температуры. И k = f(T) и D = f(T) описываются одним и тем же уравнением (уравнение Аррениуса): k = k0 exp (−E/RT), (9) где k0 − константа; Е − энергия активации химической реакции. D = D0 exp (−E1/RT), (10) 1 где D0 – константа; Е − энергия активации диффузии. Таким образом, вне зависимости от вида контролирующей стадии процесса окисления с повышением температуры скорость окисления резко возрастает. Колебания температуры, особенно переменный нагрев и охлаждение, увеличивают скорость окисления металла, так как в защитной пленке образуются трещины. Состав газовой фазы. Влияние состава газовой фазы на скорость коррозии металла велико, специфично и изменяется с изменением температуры. В частности, на скорость окисления железа и 21 стали особенно сильно влияют кислород, соединения серы, водяные пары. Приведенные ниже данные свидетельствуют о зависимости увеличения относительной скорости коррозии стали с 0,17 % углерода от состава газовой фазы при 900 оС: Состав газовой фазы Кислород Чистый воздух Чистый воздух + 2 % SO2 Чистый воздух + 5 % H2O Чистый воздух + 5 % SO2+ 5 % H2O % 200 100 118 134 276 Значительное влияние на коррозию сталей и сплавов оказывают продукты горения топлива, содержащие ванадий (например, V2O5). Это соединение находится в золе от сжигания дешевого топлива − мазута, нефтепродуктов. Зола, налипая на металл, увеличивает скорость его коррозии в десятки раз. Причина этому – «ванадиевая коррозия», обусловленная легкоплавкостью V2O5, и его способностью ОФЛЮСОВЫВАТЬ (переводить в жидкое состояние) химические соединения золы и окалины. Пятиокись ванадия участвует в процессе окисления железа: 4Fe + 3V2O5 = 2Fe2O3 + 3V2O3, V2O3 + O2 = V2O5, V2O5 + Fe2O3 = 2FeVO4. Повышение содержания СО в газовой фазе понижет скорость коррозии углеродистых и низколегированных сталей, но при больших количествах СО в газовой фазе может произойти науглероживание поверхности стали. При этом возможны следующие химические реакции: 2CO + O2 = 2CO2, 2CO = Cсаж + CO2. Давление окислителя. Если бы поверхность металла не была покрыта оксидной пленкой, то скорость окисления была бы пропорциональна парциальному давлению окислителя (для двухатомных газов – PГ2). Так как поверхность металла покрыта слоем оксида, то зависимость скорости окисления от величины парциального давления окисляющего газа определяется строением защитной пленки. В то же время при окислении железа при 700 − 950о С скорость окисления не зависит от парциального давления Po2. 22 Скорость движения газовой фазы. Окисление как гетерогенный процесс определяется скоростью подвода и отвода реагентов в зону реакции. Поэтому, чем больше скорость движения потока газа, тем больше и скорость окисления металла. Режим нагрева металла. Влияние режима нагрева металла может быть рассмотрено в зависимости от влияния колебаний температуры. Иначе говоря, переменные нагрев и охлаждение увеличивают скорость окисления ввиду нарушения сохранности защитной пленки. 2.4.3. Влияние внутренних факторов на химическую коррозию металлов ВНУТРЕННИМИ называют факторы, связанные с составом, структурой, внутренними напряжениями в металле, качеством обработки поверхности металла и др. Состав сплава. Применительно к наиболее важным конструкционным сплавам на основе железа для случая газовой коррозии можно отметить следующее. 1. При высоких температурах (более 800оС) с увеличением содержания углерода в стали скорость ее окисления и обезуглероживания уменьшается вследствие более активного образования СО, что снижает окислительный потенциал газовой фазы. 2. Сера фосфор, никель, марганец практически не влияют на скорость окисления железа. 3.Титан, медь, кобальт, бериллий заметно снижают скорость окисления железа. 4. Хром, алюминий, кремний сильно замедляют окисление железа. 5. Ванадий, вольфрам и молибден могут значительно ускорить окисление стали, которое иногда носит катастрофический характер. Структура сплава. Анализ экспериментальных данных свидетельствует о том, что чем меньше в сплаве структурных составляющих, тем выше его жаростойкость. Применительно к сплавам железо – углерод наиболее устойчивой является АУСТЕНИТНАЯ СТРУКТУРА9. Аустенит (γ-фаза) − высокотемпературная гранецентрированная модификация железа и его сплавов. В углеродистых сталях аустенит − это твѐрдый раствор внедрения, в котором атомы углерода входят внутрь элементарной ячейки γ-железа во время конечной термообработки. В сталях, содержащих другие металлы (кроме железа, легированные стали), атомы металлов замещают атомы железа в кристаллической решетке, и возникает твердый раствор замещения. Фаза названа в честь сэра Уильяма Чандлера Робертс-Остина (англ. William Chandler Roberts-Austen, 1843 − 1902). 9 23 Всего различают пять больших групп нержавеющих сталей, определяемых их микроструктурой. Наиболее распространенными являются три: АУСТЕТНИТНЫЕ (AUSTENITIC) − не магнитная сталь с основными составляющими 15−20 % хрома и 5−15 % никеля, который увеличивает сопротивление коррозии. Она хорошо подвергается тепловой обработке и сварке. Обозначается буквой A в начале наименования марки. Именно аустенитная группа сталей наиболее широко используется в промышленности и в производстве элементов крепежа. МАРТЕНСИТНЫЕ (MARTENSITIC) − значительно более твердые, чем аустетнитные стали и могут быть магнитными. Они упрочняются, закалкой и отпуском подобно простым углеродистым сталям и находят применение главным образом в изготовлении столовых приборов, режущих инструментов и общем машиностроении. Больше подвержены коррозии. Обозначаются буквой С. ФЕРРИТНЫЕ (FERRITIC) стали значительно более мягкие чем мартенситные по причине малого содержания углерода. Они также обладают магнитными свойствами. Обозначаются буквой F. Стали с двухфазной АУСТЕНИТНО−ФЕРРИТНОЙ СТРУКТУРОЙ менее устойчивы против окисления. Их меньшая жаростойкость связывается с большей неоднородностью образующейся защитной пленки, что приводит к ее разрушению при росте (неоднородность возникающих внутренних напряжений). Так хромоникелевые стали с однофазной аустенитной структурой более устойчивы против окисления, чем двухфазные: Х12Н12М2Т, Х12Н9Т ведут себя лучше, чем ОХ21Н5МД2Т или 1Х21Н5Т 2]. На жаростойкость10 чугунов кроме их состава влияет и структура. Существенное влияние оказывает форма графитных включений. Так, при шаровидной форме графита стойкость против окисления выше, чем при пластинчатой. Установлено, что предварительная холодная пластичная деформация несколько ускоряет окисление металла вследствие повышения запаса его энергии. Чем тщательнее обработана поверхность металла, тем меньше скорость его окислеЖаростойкость − окалиностойкость, способность металлических материалов противостоять химическому разрушению поверхности под воздействием воздушной или иных газообразных сред при высоких температурах. 10 24 ния, что обусловлено лучшей сохранностью защитных пленок на гладкой поверхности. 2.5. Некоторые случаи газовой коррозии металлов в технологических средах В химической промышленности многие технологические процессы или их определенные стадии протекают в газовой среде в условиях повышенных температур и давлений (рис.4). При температурах от 100 до 200 − 300 °С многие газы не опасны. Химическая активность газов и скорость газовой коррозии металлов сильно возрастают при температурах выше 200 − 300 °С. Так, Cl2 начинает действовать на железные сплавы при Рис. 4. Некоторые случаи газовой температуре > 200°C, HCl - > korozja metali w procesie technologicznym - 300°C, SO2, NO2, pary siarki. > 500°C. Te cechy zachowania środowisk gazów technologicznych i ich szerokie zastosowanie w przemyśle wymagają bardziej szczegółowego rozważenia zachowania metali w warunkach rzeczywistych. 2.5.1. Korozja żelaza, żeliwa i stali w atmosferze oparów O2, CO2, H2O. Oddziaływanie tlenu z powietrza w wysokich temperaturach na stopy żelazowo-węglowe prowadzi do utleniania żelaza z powstawaniem kamienia kotłowego, odwęgleniem stali i narostem. z żeliwa. W wyniku utleniania żelaza w wysokich temperaturach tworzy się warstwa produktów korozji zwana SKAŁĄ. Zgorzelina ma złożoną strukturę i zawiera kilka tlenków: Fe3O4 – MAGNETYT ma złożoną sieć krystaliczną spinelową; Fe2O3 – HEMATYT ma siatkę romboedryczną; FeO – VUSTIT ma wadliwą strukturę sieci krystalicznej. Wustyt powstaje w temperaturze powyżej 575 oC i przy powolnym chłodzeniu rozkłada się: 4FeO Fe3O4 + Fe. 25 Poniżej 575°C wustytu nie ma w zgorzelinie, a bezpośrednio do powierzchni stali przylega warstwa Fe3O4. Szybkość utleniania stali (patrz rozdział 2.4.1) rośnie wraz ze wzrostem temperatury zgodnie z prawem bliskim wykładniczemu (rys. 5), a we współrzędnych logk − 1/T wyraża się linią przerywaną, której każde przerwanie odpowiada jakaś transformacja. I tak, w temperaturze 575°C w zgorzelinie pojawia się podwarstwa wustytu FeO, która nie zakłóca dyfuzji tlenu, w wyniku czego zwiększa się energia aktywacji procesu i zwiększa się szybkość utleniania metalu. Przerwa w krzywej w temperaturze ~ 900 ºС odpowiada przemianie alotropowej stali. Charakter zmiany zależności temperaturowej szybkości utleniania stali wskazuje, że struktura austenityczna jest bardziej żaroodporna, w której obserwuje się wolniejszy wzrost szybkości utleniania wraz ze wzrostem temperatury. Wraz z utlenianiem w stalach i żeliwie następuje proces dekarbonizacji - zubożenie warstwy powierzchniowej w węglu na skutek oddziaływania zawartego w nich węglika żelaza z tlenem i odczynnikami zawierającymi tlen Fe3C+O2 → 3Fe+CO2 Fe3C+CO2 →3Fe+2CO Fe3C+H2O →3Fe+CO + H2. 26 Skład środowiska gazowego ma duży wpływ na szybkość utleniania stali i żeliwa. W przypadku korozji gazowej agresywnymi środkami mogą być np. chlor, związki siarki, tlen, powietrze, związki jodu itp. Ich agresywność w stosunku do różnych metali nie jest taka sama, dlatego szybkość korozji jest różna. Zatem stopień utleniania Fe, Co, Ni w temperaturze 900 °C wzrasta w kolejności H2O → CO2 → O2 → SO2. W tym przypadku metale, w zależności od szybkości korozji w atmosferze tych odczynników, układają się w rosnący szereg: Ni → Co → Fe. Jeżeli założymy, że szybkość korozji w powietrzu w temperaturze 900°C wynosi 100%, to dodatek 2% SO2 zwiększa tę szybkość do 118%, 5% H2O do 134%, 2% SO2 + 5% H2O do 276%. 2.5.2. Korozja pod wpływem produktów spalania paliw Produkty spalania paliw (węgiel, olej opałowy itp.) w większości przypadków zawierają znaczne ilości związków siarki (SO2, H2S) oraz wanadu w postaci V2O5, który jest silnym utleniaczem. Niskotopliwy tlenek wanadu oddziałuje z ochronną warstwą kamienia na powierzchni metalu, niszcząc ją i tworząc wanadany, które tworzą eutektykę11 o niskiej temperaturze topnienia Fe2O3 + V2O5 → 2FeVO4 z tlenkiem wanadu V2O5. Zatem wanadan żelaza (FeVO4) ma temperaturę topnienia = 840°C, a temperatura topnienia jego eutektyki z V2O5 wynosi 625°C; Temperatura topnienia wanadanu chromu CrVO4 wynosi 810°C, a jego eutektyka z V2O5 wynosi 665°C. Następnie wanadan aktywnie uczestniczy w procesie utleniania samego metalu 6FeVO4+4Fe → 5Fe2O3 + 3V2O3 4Fe+ 3V2O5 → 2Fe2O3 + 3V2O3 V2O3+O2 → V2O5, a sam V2O5 praktycznie się nie zużywa. Korozja wanadu w największym stopniu wpływa na wężownice, podpory i przegrody pieców pracujące w zakresie temperatur 620 – 700°C. Eutektyka (z greckiego éutektos – łatwo topiący się), układ ciekły (roztwór lub stop), będący pod danym ciśnieniem w równowadze z fazami stałymi, których liczba jest równa liczbie składników układu. 11 27 Jednocześnie szybkość korozji nawet stali chromowych i chromowo-niklowych, które nie są podatne na korozję wysokotemperaturową siarkowodorem, sięga 15 mm/rok. Pod wpływem związków siarki stale żelazowo-węglowe ulegają intensywnej korozji międzykrystalicznej na skutek większej liczby defektów w sieciach krystalicznych siarczków niż tlenków. Prowadzi to do intensyfikacji procesów dyfuzyjnych. Wraz ze wzrostem zawartości tlenku węgla (II) w produktach spalania zauważalnie maleje szybkość korozji gazowej stali węglowych i niskostopowych. Jednak jego bardzo wysokie stężenie prowadzi do nawęglenia powierzchni: 3Fe+2CO →Fe3С+CO2. Stopy wysokostopowe typu X40N50 są odporne na korozję wanadu. Wprowadzenie krzemu do stali ma również pozytywny wpływ; dodanie stali stopowej z molibdenem, wolframem i wanadem ma negatywny wpływ na trwałość stali, ponieważ te pierwiastki stopowe sprzyjają12 tworzeniu się V2O5. Stopy na bazie Cu są bardzo podatne na taką korozję. Aby zmniejszyć szybkość korozji wanadu, obecnie stosuje się: − ograniczenie temperatury pracy do 650°C; − zastosowanie stopów typu X40N50 do produkcji elementów podlegających tego typu zniszczeniom; − wdmuchiwanie do paliwa pyłu dolomitowego zawierającego tlenki magnezu i glinu, które z tlenkiem wanadu tworzą związki ogniotrwałe; − ograniczenie całkowitej zawartości sodu i wanadu w paliwie do nie więcej niż 2 10–4%. 2.5.3. Korozja w środowisku chloru i chlorowodoru Zachowanie metali w środowisku gazowego chloru i chlorowodoru zasadniczo różni się od działania innych agresywnych środowisk. Wynika to z faktu, że sole chlorkowe tworzące się na powierzchni metalu mają niską temperaturę topnienia, a w niektórych przypadkach wraz ze wzrostem temperatury ulegają sublimacji13, np. Ti + 2Cl2 → TiCl4. Promowanie - (synonimy) kierowanie, promowanie, kierowanie. Sublimacja to przejście substancji ze stanu stałego do stanu gazowego z pominięciem fazy ciekłej; to samo co sublimacja. 12 13 28 Większość tych reakcji jest egzotermiczna. Szybkość usuwania ciepła okazuje się mniejsza niż szybkość samej reakcji, w wyniku czego metale zapalają się i „palą” w atmosferze chloru. Prowadzi to do znacznego lokalnego wzrostu temperatury, a powstałe chlorki topią się i rozkładają. Najbardziej odporne na chlor są stale niklowe, ołowiowe i chromowane. Temperatura zapłonu niektórych metali w atmosferze suchego chloru: brak zapłonu. Z Ti< 20 Pb 90−100 Fe 150 Cu 200 Ni > 500. W suchym chlorowodorze w temperaturze pokojowej wiele metali i stopów ma zadowalającą odporność. Wraz ze wzrostem temperatury rezystancja materiałów metalicznych stopniowo maleje do temperatury właściwej dla każdego metalu. Maksymalne wysokie temperatury dopuszczalne podczas długotrwałej pracy metali i stopów w suchym chlorze i chlorowodorze podano w tabeli. 2. Najbardziej odpornymi metalami w suchym chlorze, z wyjątkiem metali szlachetnych, są nikiel i jego stopy. Powłoki powierzchniowe powstające na stalach niklowych i chromowo-niklowych charakteryzują się niską lotnością i zadowalającymi właściwościami ochronnymi. Tabela 2 Dopuszczalne temperatury pracy niektórych metali i stopów w atmosferze chlorowodoru i chloru3] T, °C Materiał Cl2 1200 − − 550 − 100 150 300 − Platyna Złoto Wolfram Nikiel Inconel (80% Ni, 14% Cr, 6% Fe) Miedź Stal węglowa St.Z Stal nierdzewna 12Х18Н9Т Srebro 29 HCl 1200 870 600 510−600 480 100−120 260−350 450−500 230 2.5.4. Korozja wodorowa stali Korozja wodorowa może towarzyszyć wielu procesom technologicznym zachodzącym w podwyższonych temperaturach od 200°C i ciśnieniach od 300 MPa w środowiskach zawierających wodór. Warunki te odpowiadają procesom takim jak uwodornienie węgla i ropy, synteza amoniaku i metanolu itp. Obserwuje się dwa rodzaje uszkodzeń metali przez wodór - BEZKRUCHOŚĆ WODOROWA i KOROZJA WODOROWA. Często zjawiska te nakładają się na siebie. Jeśli w gazie występuje amoniak, może również nastąpić AZOTOWANIE METALI. Kiedy mieszanina azotu i wodoru wchodzi w kontakt z metalem w warunkach podwyższonej temperatury i ciśnienia, wodór cząsteczkowy dysocjuje na powierzchni metalu. Powstały wodór atomowy dyfunduje do siatki metalowej i rozpuszcza się w niej. Gdy temperatura spada z powodu zmniejszonej rozpuszczalności, wodór ma tendencję do przejścia w stan gazowy wewnątrz metalu. W tym przypadku w metalu powstają duże naprężenia, które prowadzą do nieodwracalnej kruchości. Korozja wodorowa jest wynikiem chemicznego oddziaływania wodoru ze składnikiem węglikowym stali. Zewnętrznie przejaw korozji wodorowej oznacza silny spadek wytrzymałości stali bez zauważalnego zniszczenia powierzchni. Mechanizm korozji wodorowej obejmuje następujące etapy: - w wysokich temperaturach wodór cząsteczkowy dysocjuje na atomy, które są absorbowane przez powierzchnię stali i dyfundują do jej sieci krystalicznej; − będąc aktywnym czynnikiem redukującym, wodór atomowy redukuje węglik żelaza i oddziałuje z węglem rozpuszczonym w stali poprzez nieodwracalne reakcje: Fe3C + 2H2 3Fe + CH4 C + 4H CH4 pozbawiając stal podstawy wzmacniającej. Ponieważ procesy dyfuzyjne, w tym ruch wodoru, najłatwiej realizować wzdłuż granic ziaren, gdzie przeważają płyty cementytu, ich zniszczenie przez wodór prowadzi do zakłócenia połączenia między krystalitami, a co za tym idzie, do zmniejszenia ciągliwości stali . 30 Powstały metan ma duży rozmiar cząsteczkowy w porównaniu z parametrami sieci krystalicznej ferrytu. W rezultacie nie może on dyfundować z większości metalu i gromadzi się w jego mikrowgłębieniach i defektach, powodując wysokie ciśnienie wewnątrz wnęki i prowadząc do pęknięć. W tym przypadku pęknięcia powstają wzdłuż granic ziaren. W stalach miękkich o niskich właściwościach wytrzymałościowych metan gromadzi się głównie w warstwie przypowierzchniowej, tworząc wyraźnie widoczne na powierzchni stali spęcznienia. Oprócz udziału w tworzeniu metanu wodór ma dobrą rozpuszczalność w metalu. Stężenie wodoru rozpuszczonego w metalu zależy od ciśnienia cząstkowego wodoru na granicy faz metal-gaz i można je wyznaczyć ze wzoru: = Ks p1/2, gdzie to ilość rozpuszczonego wodoru w stali, cm3/ 100 gramów; p – ciśnienie cząstkowe wodoru, w; Ks – rozpuszczalność wodoru w stali przy p = 1 at. Ponieważ rozpuszczanie wodoru przez metal jest procesem endotermicznym, rozpuszczalność wodoru wzrasta wraz ze wzrostem temperatury. Zjawisko to nazywa się PRZEPUSZCZALNOŚCIĄ WODORU (VH) 4]. Przepuszczalność wodoru przez stal zależy od zawartości w niej węgla i pierwiastków stopowych i maleje wraz ze wzrostem zawartości węgla, o czym świadczą dane podane w tabeli. 3. Tablica 3 Wpływ zawartości węgla na przepuszczalność wodoru stali węglowych Metal C,% wag. VН, cm3/cm2h mm–1 Żelazo techniczne 0,04 5,10 Stal 15 0,15 4,09 Stal 30 0,27 2,79 Stal U10A 1,10 2,24 Korozja wodorowa nie występuje natychmiast od momentu wystawienia stali na działanie wodoru, ale po pewnym czasie. Ten czas, w którym nie zachodzą zmiany w mikrostrukturze i właściwościach mechanicznych stali, nazywany jest OKRESEM INKUBACJI PROCESU ODwęglania Stali. 31 Okres inkubacji ma ogromne znaczenie praktyczne, gdyż w zasadzie od niego zależy bezpieczny czas pracy sprzętu. Okres inkubacji zależy głównie od temperatury i ciśnienia. Wraz ze wzrostem temperatury i ciśnienia okres inkubacji maleje. Jednocześnie na czas trwania okresu inkubacji duży wpływ ma skład chemiczny stali. Temperatura i ciśnienie cząstkowe wodoru wpływają nie tylko na czas trwania okresu inkubacji, ale także na szybkość odwęglenia stali podczas korozji wodorowej. 2.5.5. Korozja karbonylowa Korozja karbonylowa zachodzi w procesach technologicznych z udziałem węgla pod podwyższonymi ciśnieniami i temperaturami. Do takich procesów zalicza się np. produkcję alkoholi metylowych i butylowych, konwersję metanu i tlenku węgla. W normalnych warunkach CO jest obojętny w stosunku do metali. W wysokich temperaturach i ciśnieniach tlenek węgla reaguje z wieloma metalami i tworzy karbonylki. Na przykład Fe + nCO = Fe(CO)n. Żelazo i CO mogą tworzyć trzy związki: tetrakarbonyl - Fe(CO)4, pentakarbonyl - Fe(CO)5 i nanokarbonyl - Fe(CO)g. Wszystkie te związki są dość niestabilne i rozkładają się wraz ze wzrostem temperatury. Najbardziej stabilnym związkiem spośród nich jest Fe(CO)5. Tworzenie karbonylków wzrasta wraz ze wzrostem temperatury od 100 şС, następnie w zakresie temperatur 140 – 180 şС obserwuje się ich prawie całkowitą dysocjację na Fe i CO. Tlenek węgla (II) może tworzyć podobne związki z wieloma innymi metalami. Korozja karbonylowa powoduje zniszczenie i rozluźnienie wierzchniej warstwy metalu do głębokości 5 mm. Zmiany w strukturze metalu w większej odległości od powierzchni już nie występują. 2.5.6. Korozja w nieelektrolitach NIEELEKTROLITY to ciecze, które nie przewodzą prądu elektrycznego. Ciekłe media żrące pochodzenia nieorganicznego - ciekły brom, stopiona siarka itp. Ciekłe substancje organiczne - benzen, chloroform itp., Paliwa ciekłe (olej, nafta, benzyna itp.), Oleje smarowe. W czystej postaci są mało agresywne, jednak obecność w nich nawet niewielkich ilości zanieczyszczeń (merkaptanów14, siarkowodoru, wody, tlenu itp.) gwałtownie zwiększa ich aktywność chemiczną. Zatem merkaptany i siarkowodór zawarte w ropie naftowej powodują korozję Fe, Cu, Ni, Ag, Pb, Sn z utworzeniem ich odpowiednio merkaptydów (Me−(S−R)n) i polisiarczków (Me2Sx). Ślady wody w czterochlorku węgla zwiększają jego korozyjność na skutek tworzenia się składników przewodzących (HCl) w wyniku hydrolizy i powstawania korozji na drodze mechanizmu elektrochemicznego: CCl4 + H2O → COCl2 + 2HCl CCl4 + 2H2O + kаt → CO2 + 4HCl Korozja metali w nieelektrolitach przebiega głównie poprzez mechanizm chemiczny. Proces ten składa się z kilku etapów, z których każdy określa szybkość korozji: 1) dyfuzja utleniacza do powierzchni metalu; 2) chemisorpcja środka utleniającego; 3) reakcja chemiczna; 4) desorpcja produktów korozji z powierzchni metalu; 5) dyfuzja produktów korozji do objętości nieelektrolitu. W niektórych przypadkach na powierzchni metalu z produktów korozji tworzy się film ochronny, co prowadzi do zahamowania procesu korozji ze względu na trudność dyfuzji utleniacza do powierzchni metalu. W zależności od właściwości ochronnych powstałej folii i jej rozpuszczalności można zastosować kontrolę dyfuzyjną, kinetyczną lub mieszaną dyfuzyjno-kinetyczną. Temperatura znacząco wpływa na szybkość korozji chemicznej metali w nieelektrolitach (patrz rozdział 2.4.). Wpływ temperatury na szybkość procesu jest w niektórych przypadkach skomplikowany przez zmiany rozpuszczalności utleniacza i warstwy produktów korozji metalu w nieelektrolicie, gdy zmienia się temperatura. 14 Merkaptany to wodorosiarczkowe pochodne węglowodorów: R – S –H. 33 Obecność wody w nieelektrolitach w znaczący sposób aktywuje korozyjne działanie zanieczyszczeń i powoduje intensywną korozję elektrochemiczną metali, tj. zmienia się mechanizm procesu korozji. Aby zabezpieczyć metale w nieelektrolitach przed korozją chemiczną, wybiera się metale i stopy stabilne w danym środowisku (np. aluminium i jego stopy są stabilne w benzynie krakowanej) i nakłada się powłoki ochronne (np. stal w środowisko siarkowodoru jest pokryte aluminium). 2.6. Metody ochrony metali przed różnymi rodzajami korozji gazowej 2.6.1. Metody zabezpieczania stali przed korozją gazową Do zabezpieczenia stali przed korozją gazową najczęściej stosuje się żaroodporne POWŁOKI ALOJOWE i POWŁOKI OCHRONNE. Stopowanie - (niem. legieren – „łączyć”, od łac. ligare – „wiązać”) – dodawanie zanieczyszczeń do składu materiałów w celu zmiany (poprawy) właściwości fizycznych i/lub chemicznych materiału bazowego. Główne metody ochrony metali przed utlenianiem w wysokich temperaturach opierają się na STOPACH. Odporność metali utlenionych w wysokich temperaturach zależy od właściwości ochronnych warstwy tlenkowej pokrywającej metal. Pierwiastkami tworzącymi warstwę ochronną na stopach żelaza z węglem są chrom, aluminium i krzem. Pierwiastki te utleniają się łatwiej w wysokich temperaturach w powietrzu niż metal stopowy i tworzą bardziej stabilną zgorzelinę. Zgodnie z teorią stopowania żaroodpornego składnik stopowy tworzy na powierzchni metalu warstwę ochronną składającą się wyłącznie z tlenku składnika stopowego. Pierwiastek stopowy musi spełniać następujące podstawowe wymagania: Tlenek powstający na powierzchni metalu musi mieć charakter ciągły, tj. stosunek Vok/VMe musi wynosić >1. Warstwa tlenku musi mieć wysoką rezystancję omową rMeO > rFe, wielkość jonów składnika stopowego musi być mniejsza niż wielkość jonów metali nieszlachetnych, tj. rMe< rFe. Это условие вытекает из следующих соображений: 34 а) меньший радиус иона легирующего компонента по сравнению с ионом основного металла позволяет предполагать у легирующего компонента больший коэффициент диффузии в сплаве, что может обеспечить преимущественный выход ионов компонента на поверхность сплава и, следовательно, преимущественное образование оксида легирующего компонента; б) меньший радиус иона легирующего компонента позволит образовать оксид с соответствующими меньшими параметрами, который будет оказывать большое сопротивление диффузии через него и, следовательно, создавать большое затруднение для окисления основного металла. Это подтверждается сравнением размеров ионных радиусов железа и легирующих элементов: хрома, алюминия и кремния Ион металла Ионный радиус, Аº Fe2+ 0,74 Cr6+ 0,52 Al3+ 0,50 Si4+ 0,41. А энергия образования оксида легирующего компонента должна быть больше энергии образования оксида основного металла ΔGMeO<ΔGFeO. Если это условие не соблюдается, то оксид легирующего компонента будет восстанавливаться основным компонентом, как например, оксидная пленка меди, которая не может быть устойчивой на железном сплаве вследствие возможности протекания реакции CuO+Fe → FeO+Cu. При этом необходимо соблюдение условий: высокая температура плавления оксида легирующего компонента, низкая упругость диссоциации, а также отсутствие низкоплавких эвтектик в смеси с другими оксидами для того, чтобы оксид легирующего компонента был достаточно устойчив. В качестве примера можно привести элемент бор, который является аналогом Al по таблице Менделеева, но дает легкоплавкие оксиды. Температура плавления ВО3 − 294 0С, и поэтому бор, в отличие от алюминия, не может быть легирующим компонентом, повышающим жаростойкость. Необходимым условием является также, образование твердых растворов между легирующими компонентами и основным металлом, так как при этом условии возможно равномерное распределение этого элемента на поверхности и образование сплошной пленки оксида легирующего компонента. 35 Существует и другая теория, согласно которой легирующий элемент образует на поверхности сплава смешанные оксиды, обладающие повышенными защитными свойствами, по сравнению с оксидами чистых компонентов. Механизм повышения жаростойкости можно свести к тому, что легирующий компонент должен уменьшить возможность образования в окалине вюститной фазы (FeO) на стали и, таким образом, благоприятствовать образованию шпинельной фазы типа Fe3O4ּγFe2O3 с возможно меньшим параметром решетки. Жаростойкие легированные стали имеют на поверхности оксидные слои со структурой именно шпинели. Еще более высокими защитными свойствами обладают сложные шпинели типа FeOּMe2O3 или Fe2O3 ּMeO. С целью экономии легированных сталей жаростойкость углеродистых сталей повышают, применяя защитные покрытия. Для нанесения на сталь защитных покрытий пользуются следующими методами: ТЕРМОДИФФУЗИОННЫМ, НАПЛАВКОЙ И ПЛАКИРОВАНИЕМ. Для нанесения покрытий термодиффузионным методом стальное изделие после очистки поверхности механическим способом и травлением помещают в реакционную смесь, состоящую из порошка наносимого металла и катализатора. Например, при нанесении алюминиевых покрытий смесь содержит измельченный ферроалюминиевый и хлористый аммоний. Процесс насыщения поверхности стали алюминием (АЛИТИРОВАНИЕ) производится при высоких температурах (900−9500С), в результате чего происходит разложение хлористого аммония на аммиак и хлористый водород: NH4Cl → NH3 + HCl, который, в свою очередь, взаимодействует с алюминием 2Al +6HCl → 3H2 + 2AlCl3. На поверхности стального изделия происходит выделение атомарного алюминия AlCl3 +Fe → FeCl3 + Al, который диффундирует в сталь. Значительное повышение жаростойкости стали термодиффузионным покрытием обусловлено образованием окислов Al2O3 или двойных окислов FeAl2O4 в алитированном слое. На поверхности хромированных или силицированных изделий образуются соответствующие оксиды хрома или кремния. 36 НАПЛАВКА − это нанесение слоя металла или сплава на поверх- ность изделия посредством сварки плавлением. ПЛАКИРОВА́НИЕ − (фр. plaquer − накладывать, покрывать) термомеханическое покрытие − нанесение на поверхность металлических листов, плит, проволоки, труб тонкого слоя другого металла или сплава термомеханическим способом. Для защиты от карбонильной коррозии применяют хромистые стали с содержанием 30 % Сг, хромоникелевые стали с содержанием 23 % Сг и 20 % № и марганцевые бронзы доя работы при температуре до 700 °С и давлении до 35 МПа. При более низких параметрах возможно применение менее легированных сталей типа Х18Н9. Сырьем для синтеза мочевины CO(NH2)2 является NH3 и СО2. Процесс протекает при температуре 175−190°С и давлении 20 МПа. Хромистые нержавеющие стали различных марок непригодны для изготовления основных аппаратов. Наибольшую стойкость имеют стали, легированные молибденом, и хромникельмолибденовомедные стали. Важным фактором для повышения коррозионной устойчивости является тщательная очистка газов от сероводорода и дополнительное введение в систему кислорода в количестве 0,5- 1,0 об.% от содержания СО2. При сернистой коррозии сера и ее соединения - сернистый ангидрид (SO2), сероводород (H2S), меркаптаны или тиоспирты и т.д. являются достаточно агрессивными, коррозионно−активными веществами. Наиболее активным компонентом при высокотемпературной газовой коррозии является сероводород. Он даже более опасен, чем диоксид серы. Сернистый газ SO2 является исходным продуктом при производстве серной кислоты. Его получают при обжиге серного колчедана, сжигании серы, из сероводорода при утилизации отходящих газов металлургических производств. Чугунные детали скребков конверторных печей кипящего слоя, зубья и гребки колчеданных печей, котлы−утилизаторы, сухие электрофильтры, газоходы обжиговых газов в производстве серной кислоты часто выходят из строя вследствие газовой коррозии. В результате коррозии черных металлов в сернистом газе при температурах 300°С и выше образуется слоистая окалина, состоящая из FeS, FeO и др. 37 При температуре газа более 400 °С для деталей из чугуна характерно увеличение объема металла, достигающего 10 % от начальной величины. При этом резко снижается прочность материала. Детали испытывают коробление, трескаются и разрушаются. Это явление называется «ростом» чугуна и объясняется внутренним окислением металла. Максимальный рост чугуна наблюдается при 700 °С. К ростоустойчивым чугунам относятся высоколегированные хромистые чугуны, карбидный чугун типа «пирофераль» и «чугаль» Сернистый газ при высоких температурах окисляет никель. При этом образуется окалина, в состав которой входят NiS и NiO: 3Ni + S02 = NiS + 2NiO. Сернистый никель образуется и при действии на металл сероводорода: Ni + H2S = NiS + Н2. Сульфид никеля с металлическим никелем образует легкоплавкую эвтектику с температурой плавления около 625 °С. Образование этой эвтектики в сталях, содержащих никель, происходит преимущественно по границам зерен, вызывая разрушение металла. Стали с содержанием никеля выше 15 % очень чувствительны к действию сернистого газа. В процессе окисления они теряют механическую прочность. Поэтому при работе с газовой средой, содержащей диоксид серы, при температурах до 400 °С используют углеродистые стали, а при более высоких температурах − хромистые стали. Наиболее употребительны жаростойкие стали − 4Х9СА, Х6СЮ, XI7, ОХ 17 Г, X1800, Х25Т. Интенсивное образование окалины происходит при температурах выше 800−1000 °С. К жаропрочным сталям в этой среде относятся Х5М, Х6СМ, XI8Н12Т, Х23Н18. Рабочая температура для этих сплавов 550−600 °С (для Х23Н18 − 1000 °С). Сухой сернистый газ реагирует с алюминием очень медленно. Поэтому алюминий используют для защиты от коррозии деталей и узлов теплообменников и контактных аппаратов. Сухой сероводород при комнатной температуре не представляет опасности для обычных углеродистых сталей. С повышением температуры опасность сероводородной коррозии углеродистых сталей значительно увеличивается. При температуре выше 300 °С 38 железо подвергается сильной коррозии в серосодержащих газовых средах. Легирование хромом в количестве > 12% zwiększa odporność na korozję w temperaturach do 700-800 °C. Podczas korozji stali chromowej tworzy się zgorzelina, której zewnętrzna warstwa składa się z siarczku żelaza. W tej warstwie praktycznie nie ma chromu. Cały utleniony chrom koncentruje się w warstwie wewnętrznej, która ma właściwości ochronne. Ferrytyczne Rys. 5 10 15 20 mają dobrą odporność chemiczną w atmosferze siarki-Cr,% wag. wodoru. 6. Szybkość korozji stopów chromu zawierających 25–30% stałej stali w parach oleju w temperaturze 650 °C. Liczby na krzywych wskazują chrom. Zawartość H2S, % Szczególnie niebezpieczna jest łączona obecność związków siarki i innych składników żrących. Zatem w przemyśle naftowym podczas termicznej obróbki olejów siarkowych szczególne zagrożenie stwarza mieszanina siarkowodoru i wodoru. Z tych pokazanych na ryc. 6 dane pokazują, że szybkość korozji stali chromowych wzrasta wraz ze wzrostem stężenia siarkowodoru w parach oleju. W tym przypadku 10-krotny wzrost stężenia H2S powoduje ponad 12–15-krotny wzrost szybkości korozji. Podczas spalania paliwa tworzą się złożone mieszaniny gazów zawierające O2 i różne tlenki, w tym zanieczyszczenia siarkowe. W takich przypadkach obserwuje się korozję siarczkowo-tlenkową. Folia ochronna na metalu składa się zwykle z kilku warstw. Warstwa zewnętrzna jest wzbogacona w tlen i składa się z tlenku metalu, natomiast warstwy wewnętrzne przylegające do powierzchni metalu zawierają zwiększone ilości siarki i siarczków. Jeśli podczas spalania paliwa powstaje popiół zawierający tlenek wanadu V2O5, wówczas szybkość korozji wzrasta bardzo szybko. Przyczyny korozji stali wanadowej omówiono wcześniej (patrz rozdział 2.5.2). 39 Stale chromowe zawierające 4-6% Cr są uważane za półżaroodporne. Stale tej klasy, ze względu na swoją dostępność, zwiększoną odporność korozyjną i wytrzymałość, znajdują szerokie zastosowanie w przemyśle naftowym do produkcji jednostek krakingowych. Wytrzymałość cieplna tych stali w powietrzu i spalinach ze znaczną zawartością związków siarki w temperaturach 500-600 ºС jest około 3 razy większa niż odporność cieplna stali niestopowych. 2.6.2. Metody ochrony przed korozją wodorową Jednym z głównych sposobów zwiększenia wodoroodporności stali jest STOPOWANIE ich mocnymi pierwiastkami węglikotwórczymi, które tworzą bardziej stabilne węgliki niż cementyt. Takimi pierwiastkami są chrom, wolfram, molibden, wanad, niob, tytan. Rodzaj fazy węglikowej ma duży wpływ na odporność stali na wodór. W stalach stopowych z chromem odporność na wodór maleje w zależności od składu fazy węglikowej w następujący sposób: (Cr,Fe)23C6(Cr,Fe)23C6 + (Cr,Fe)7C3(Cr,Fe) 7C3 (Cr,Fe)7C3 + (Fe,Cr)3C(Fe,Cr)3C Z powyższego szeregu wynika, że największą odporność na wodór w stalach chromowych osiąga się przy tworzeniu węglików (Cr,Fe )Typ 23C6. Tworzenie się tego rodzaju węglika o zawartości węgla w stali wynoszącej 0,05% następuje, gdy w stali znajduje się co najmniej 6% chromu, a wraz ze wzrostem zawartości węgla konieczne jest zwiększenie zawartości chromu. W przypadku stopowania stali z pierwiastkami tworzącymi węgliki silniejsze niż chrom, największą odporność stali na wodór osiąga się, gdy pierwiastki te są zawarte w ilościach wystarczających do związania całego węgla w węgliki typu MeC. W przypadkach, gdy elementy urządzeń pracują w warunkach dużych obciążeń mechanicznych, stale wodoroodporne muszą posiadać także odporność cieplną. Do tych warunków stosuje się stale o zawartości 3% Cr, dodatkowo stopowe Mo, V lub W, np. stal w gatunku 20Х3МВФ (EI579). Gatunki stali X5M, X5VF, X9M, 1X13 mają jeszcze większą odporność. W najcięższych warunkach stosuje się austetyczną żaroodporną stal chromowo-niklową 0Х18Н10Т lub Х17Н17М2Т. 40 Inną powszechnie stosowaną metodą zwiększania wodoroodporności stali jest POKŁADANIE ich metalami, które mają niższą przepuszczalność wodoru niż metal nieszlachetny. Umożliwia to zmniejszenie stężenia wodoru na granicy faz metalu nieszlachetnego z warstwą okładzinową i zmniejszenie jego agresywnego działania na metal nieszlachetny. Obliczenia efektywnego ciśnienia wodoru na granicy faz baza-metal powłoki dokonuje się według równania: 1/2 1/2 p2 = p1 (11) + gdzie p1 to ciśnienie wodoru na zewnętrznej powierzchni powłoki warstwa okładzinowa; p2 to efektywne ciśnienie wodoru na granicy faz powłoki i metalu nieszlachetnego; = VН1/VН2 to stosunek przepuszczalności wodoru odpowiednio warstwy okładzinowej i metalu nieszlachetnego; = l1/l2 – stosunek grubości odpowiednio warstwy okładziny do materiału rodzimego. W celu zwiększenia przepuszczalności wodoru metale i stopy można ułożyć w rzędzie: aluminium, miedź, nikiel, X18N10T, 0X13. W praktyce w przemyśle chemicznym i rafinacji ropy naftowej stosuje się głównie bimetale wykonane ze stali węglowych lub niskostopowych z warstwą ochronną ze stali 0Х13 lub Х18Н10Т. Temperatura pracy urządzeń wykonanych z wybranej stali powinna być utrzymywana o 25°C poniżej temperatury, przy której możliwa jest korozja wodorowa. Rozdział 3. KOROZJA ELEKTROCHEMICZNA METALI 3.1. Potencjały elektrod metali Po zanurzeniu metalu w elektrolicie następuje skok potencjału na granicy faz w wyniku utworzenia podwójnej warstwy elektrycznej 5, 6. Jeśli metalową płytkę, na przykład miedzianą, zanurzymy w wodzie lub roztworze soli miedzi, wówczas dodatnio naładowane jony Cu2+ zaczną przemieszczać się z warstwy metalu znajdującej się na granicy z wodą do wody. W tym przypadku w sieci krystalicznej metalu pojawia się nadmiar elektronów, a płyta zyskuje ładunek ujemny. Pomiędzy ujemnie naładowaną płytką a jonami dodatnimi, które przeszły do roztworu, powstaje przyciąganie elektrostatyczne, co zapobiega dalszemu przejściu jonów miedzi do roztworu, tj. Procesowi rozpuszczania metalowych przystanków. Jednocześnie rozwija się proces odwrotny: jony miedzi z roztworu zbliżając się do powierzchni płytki, przyjmują z niej elektrony i przechodzą w stan neutralny. Po pewnym czasie ustala się stan równowagi dynamicznej, w którym szybkość przejścia jonów z metalu do roztworu jest równa szybkości wyładowania jonów z roztworu na metal. Opisane zjawisko pokazano schematycznie na rys. 7 (dla uproszczenia jony metali pokazano w postaci nieuwodnionej). Równowaga pomiędzy jonami w roztworze i metalem w rozważanym przykładzie jest wyrażona równaniem: Cu2+р−р + 2ē Cućkryształ W równaniu równowagi reakcji elektrochemicznej elektrony są zwykle zapisywane po lewej stronie, tj. proces redukcji jest zapisany. Ryc.7. Schemat wyglądu podwójnej warstwy elektrycznej na granicy faz metal-roztwór Jak widać z powyższego przykładu, gdy metal wchodzi w kontakt z roztworem swojej soli, dwie stykające się fazy uzyskują przeciwne ładunki. W rezultacie na granicy faz powstaje podwójna warstwa elektryczna i następuje skok potencjału () pomiędzy metalem a roztworem. Jeśli do roztworu opuści się dwie płytki z różnych metali (M1 i M11), powstanie para galwaniczna, w której mniej aktywny metal zostanie zredukowany, a bardziej aktywny utleniony (ryc. 8). Ryż. 8. Schemat działania ogniwa galwanicznego Ogólnie rzecz biorąc, praca tej pary galwanicznej jest zdeterminowana różnicą potencjałów (sem ogniwa galwanicznego) i towarzyszy jej bilans reakcji połówkowych: E = ox − red, gdzie ox jest potencjałem utleniacza, V; czerwony – potencjał środka redukującego, V. Men+(wół) + ne ⇆ Меº Me°(czerwony) − ne ⇄ Меn+. Kiedy liczba kationów przechodzących do roztworu w jednostce czasu zrówna się z liczbą kationów osadzonych na powierzchni metalu, nastąpi równowaga dynamiczna i proces rozpuszczania zostanie zatrzymany. Dlatego przejście dużej liczby atomów jonów metali do roztworu w takich warunkach jest niemożliwe. Jeśli jednak równowaga elektrycznej warstwy podwójnej zostanie zakłócona przez wyładowanie elektronów lub usunięcie atomów jonów metali, proces korozji będzie przebiegał bez przeszkód. Potencjały elektrod metali, które są w równowadze z własnymi jonami, nazywane są równowagą. Wartość potencjału równowagowego można obliczyć dla dowolnej aktywności jonu, korzystając z równania Nernsta: E = E° + (RT /nF)ln(aMen+), (12) n+ gdzie E° jest standardową różnicą potencjałów przy aMe = 1; R – uniwersalna stała gazowa, 8,3 kJ/kmolK; T – temperatura bezwzględna, °K; n – wartościowość metalu; F – liczba Faradaya 96500 C/mol; 43 aМen+ – aktywność jonów metali w mol/l. Jeśli podstawimy wszystkie stałe w temperaturze 25°C (T = 298 K) i pomnożymy przez 2,3, aby przejść od logarytmów naturalnych do dziesiętnych, otrzymamy następujące wyrażenie E = E° + (0,0592/n)log(aMen+), ( 13) Gdy roztwór zostanie rozcieńczony, potencjał metalu przesuwa się na stronę ujemną. Jeśli np. aktywność jonów Zn2+ w roztworze soli cynku będzie wynosić 10–2 mol/l, to potencjał równowagowy cynku zanurzonego w tym roztworze będzie równy: E = − 0,76 + (0,0592/2) lg10−2 = − 0,819 V, (14) Wraz ze wzrostem stężenia jonów metali w roztworze potencjał metalu natomiast przesuwa się w kierunku dodatnim. Zatem jeśli przyjąć aktywność jonów cynku jako 10 mol/l, to potencjał cynku wyniesie E = − 0,76 + (0,0592/2)log10 = − 0,73 V, (15) Potencjały elektrod metali, w których The w procesie wymiany, który określa potencjał, biorą udział nie tylko własne, ale także inne jony i atomy, które nazywane są nierównowagowymi lub nieodwracalnymi. Dla potencjałów nierównowagowych wzór Nernsta nie ma zastosowania, ponieważ reakcje zachodzące na metalu, tj. utrata i nabycie elektronów następuje na różne sposoby, a potencjał nie może scharakteryzować początku równowagi żadnej reakcji na elektrodzie. Na wielkość potencjałów nierównowagowych ma wpływ charakter elektrolitu, temperatura, ruch elektrolitu, stężenie roztworu itp. Przykładowo potencjał glinu w 3% NaCl wynosi – 0,6 V, w 0,05 M Na2SO4 – 0,47 V, oraz w 0,05 M Na2SO4 + H2S - 0,23 V. Ze względu na elektrochemiczną niejednorodność powierzchni metalu lub elektrolitu na metalu tworzą się obszary o różnych potencjałach. Głównymi przyczynami powodującymi elektrochemiczną niejednorodność powierzchni metalu mogą być: - LIKWACJA - (La liquation, Saigerung) - to zdolność stopów do rozpadu podczas przejścia ze stanu ciekłego do stałego na składniki lub poszczególne związki o różnych temperaturach topnienia ; − niejednorodność folii ochronnej; 44 – niejednorodność warunków fizycznych – nierówna temperatura w różnych obszarach metalu, obecność obszarów odkształconych, koncentratory naprężeń; − niejednorodność elektrolitów – różnica w stężeniu tlenu lub soli, różna wartość pH. Obszary o bardziej ujemnym potencjale nazywane są ANODYCZNYMI, obszary o bardziej dodatnim potencjale nazywane są KATODĄ. Stopień niejednorodności powierzchni metalu zależy od różnicy potencjałów sekcji katodowej i anodowej. 3.2. Termodynamika korozji elektrochemicznej O możliwości samoistnego wystąpienia procesu korozji decyduje spadek energii swobodnej (potencjału izobaryczno-izotermicznego) G< 0: G = −nFE < 0, (16) где n – эквивалентное число электронов; Е = К – А – эдс гальванического элемента, образующегося при контакте металла с окислителем в процессе коррозии; К – обратимый потенциал катодной реакции (окислителя); А – обратимый потенциал анодной реакции (металла – восстановителя); F – число Фарадея. Принципиальная возможность протекания процесса электрохимической коррозии металла имеет место при К > A lub E > 0. Odwracalny potencjał redoks środka utleniającego w elektrolicie musi być w tych warunkach bardziej dodatni niż odwracalny potencjał metalu w tych samych warunkach. Do ustalenia granic termodynamicznej możliwości korozji elektrochemicznej metali można zastosować diagramy Pourbaixa. Wykresy stanowią graficzne przedstawienie zależności potencjałów elektrod odwracalnych (w woltach w skali wodorowej) od pH roztworu dla stanów równowagi: z udziałem elektronów – linie poziome; z udziałem elektronów i jonów H+ lub OH– - linie ukośne; z udziałem jonów H+ i OH–, ale bez udziału elektronów (wartość pH tworzenia hydratu) – linie pionowe. 45 Ryc.9. Wykres E – pH dla układu Al-H2O w temperaturze 25°C Przykład na rys. Rysunek 9 przedstawia diagram Pourbaix dla układu Al – H2O. Każdy obszar diagramu odpowiada jednemu stanowi stabilnemu termodynamicznie: stanowi metalicznemu w postaci jonów w roztworze, w postaci tlenków lub wodorotlenków. Ponieważ ustalenie równowagi w roztworze zależy nie tylko od jonów wodoru, ale także od innych jonów, granicę obszarów tworzy kilka linii równowagi. Każda z tych linii odpowiada określonej aktywności odpowiednich jonów. W obszarze na dole diagramu metaliczny aluminium jest stabilny termodynamicznie i nie koroduje. W lewej części nad linią poziomą stan glinu w postaci jonów Al3+ w roztworze jest stabilny termodynamicznie. Środkowy obszar odpowiada stabilności faz stałych tlenku Al2O3 lub wodorotlenku Al(OH)3, które w procesie korozji tworzą warstwę ochronną na powierzchni aluminium. Prawa strona charakteryzuje warunki, w jakich aluminium będzie ulegało korozji z utworzeniem w roztworze anionu AlO2–. 3.3. Mechanizm korozji elektrochemicznej Korozja metali w elektrolitach zachodzi w wyniku tworzenia się par galwanicznych, zwanych parami korozyjnymi. Jeśli opuścimy odpowiednio płytki cynkowe i żelazne do naczynia z roztworami chlorków cynku i żelaza (podobnie jak na wykresie pH me na ryc. 8) i połączymy je przewodem zewnętrznym, to podłączony do obwodu miliamperomierz pokaże obecność prądu elektrycznego w obwodzie. Tworzy się para galwaniczna, w której cynk utlenia się i przechodzi do roztworu (anoda). Żelazo nie utlenia się i nie przechodzi do roztworu, ponieważ w połączeniu z cynkiem działa jak katoda. Żelazo nie połączone z cynkiem koroduje w podobnym roztworze. W praktyce stopy zawierają dużą liczbę mikroelementów i różnych metali. Innymi słowy, różne metale znajdują się w tej samej płaszczyźnie w bezpośrednim kontakcie ze sobą i z elektrolitem. Rezultatem jest ogniwo wieloelektrodowe, w którym anody występują na przemian z katodami. W miejscach anodowych jony metali (Men+) przechodzą do roztworu. Uwolnione elektrony (nē) przemieszczają się przez metal od anody do katody. Rozpuszczanie jonów metali w roztworach wodnych należy przedstawiać w wyniku oddziaływania jonów - atomów znajdujących się na zewnętrznej powierzchni metalu z polarnymi cząsteczkami wody. Oddziaływanie rozpuszczonych jonów metali z dipolami wody spowodowane jest siłami przyciągania elektrostatycznego. W rezultacie wokół każdego jonu w większym lub mniejszym stopniu (w zależności od wielkości jego ładunku) tworzy się otoczka dipoli wody (zjawisko hydratacji). W wyniku hydratacji zwiększa się efektywny promień jonu, w wyniku czego ruchliwość uwodnionych jonów znacznie maleje. Procesowi hydratacji towarzyszy uwalnianie energii, natomiast procesowi odwodnienia potrzeba energii. Oprócz dipoli wody jon może być pokryty powłoką innych dipoli. Mówiąc bardziej ogólnie, zjawisko to nazywa się solwatacją15. Zatem korozja elektrochemiczna składa się z następujących równolegle etapów: - proces anodowy, który polega na przejściu jonów metali z powierzchni do roztworu i ich uwodnieniu: SOLVACJA - proces tworzenia asocjatów pomiędzy rozpuszczalnikiem a rozpuszczoną substancją substancji, produktem solwatacji jest SOLVAT. Jeśli rozpuszczalnikiem jest woda, to NAWODNIENIE, produktem hydratacji jest WODNIAN. 15 47 Me° − nē Mężczyźni+ + mH2O Mężczyźni+ mH2O. hydrat jest procesem katodowym, który polega na asymilacji (wychwytywaniu) elektronów przez jakiś depolaryzator (D): D + nē . Ponieważ procesy anodowe i katodowe są niezależne i zachodzą łatwiej: anodowy w obszarach o bardziej ujemnym potencjale początkowym powierzchni, a katodowy w bardziej dodatnich - a w praktycznych warunkach niezbędna do tego jest elektrochemiczna niejednorodność powierzchni metalu Należy zauważyć, że procesy te zachodzą głównie lokalnie. Przepływ prądu elektrycznego odbywa się na metalu - poprzez ruch elektronów z obszarów anodowych do katody, a w roztworze następuje jednocześnie ruch jonów. Zatem natężenie prądu może i służy jako kryterium szybkości korozji elektrochemicznej. Oblicz zużycie materiału anody, tj. ilość skorodowanego metalu z 1 cm2 jego powierzchni można obliczyć korzystając ze wzoru Faradaya: K = Q A / F n = i A / F n, gdzie K jest ilością skorodowanego metalu metal, g/cm2 ; Q jest ilością energii elektrycznej przepływającej w czasie τ, [s] pomiędzy sekcjami anody i katody; i – gęstość prądu, A/cm2; F – liczba Faradaya; n – wartościowość metalu; A jest masą atomową metalu. 3.4. Polaryzacja procesów elektrodowych i jej przyczyny W przypadku zwarcia elektrod elementu korozyjnego następuje znaczna zmiana potencjałów elektrod, ale rezystancja pary praktycznie się nie zmienia. W tym przypadku obserwuje się spadek siły elektromotorycznej elementu korozyjnego. Zmiana potencjałów początkowych, prowadząca do zmniejszenia prądu korozji, a w konsekwencji szybkości korozji, nazywana jest POLARYZACJĄ. Zmiana potencjałów (16) elektrod podczas pracy elementu korozyjnego wskazuje, że potencjał katody staje się bardziej ujemny (POLARIZACJA KATODY), a potencjał anody staje się bardziej dodatni (POLARIZACJA ANODOWA): K = Ko – K, (17) o A = A + A, (18) gdzie K i A są wartościami potencjałów elektrod elementu roboczego; Кº i Аo – początkowe wartości potencjałów elektrod przed zamknięciem obwodu; K i A – przesunięcie potencjału (polaryzacja) katody i anody. Zatem różnica potencjałów E = K − A maleje. POLARYZACJA jest konsekwencją opóźnienia procesów elektrodowych od przepływu elektronów w elemencie zwartym. Rezystancje omowe (R) mają niewielki wpływ na zmniejszenie prądu korozji, ponieważ są zwykle małe. Duże znaczenie mają opory polaryzacyjne, które wiążą się albo z trudnościami w wyładowaniu elektronów na katodzie (Pk), albo z trudnościami w przejściu dodatnich jonów metali z sieci metalicznej do roztworu (PA). Te rezystancje, które mają wymiar omowy, są główną przyczyną spadku szybkości korozji elektrochemicznej. Natężenie prądu w chwili zamknięcia obwodu zgodnie z prawem Ohma określa się wzorem Iinit = (Кº − Аo)/R, (19). Początkowa wartość prądu korozyjnego (Iinit) jest wprost proporcjonalna do różnicy pomiędzy początkowymi wartościami potencjałów Kº i Ao, pod warunkiem, że Kº > Ao, i odwrotnie proporcjonalna do rezystancji. Prąd korozyjny w stanie ustalonym (Iogniwo robocze) jest określone równaniem: Iogniwo robocze = (K – A)/(R + PA + PK). Zatem pracuję. el-ta< Iнач. Снижению скорости электрохимической коррозии или уменьшению коррозионного тока могут способствовать: − уменьшение степени термодинамической стабильности, т. е. сближение равновесных потенциалов анодного Аº и катодного Кº процессов; − торможение катодных процессов, иначе говоря, увеличение катодной поляризуемости РК; 49 − торможение анодных процессов, то есть увеличение анодной поляризуемости РА. Поляризация является весьма желательным явлением в коррозионных процессах, так как электроды замкнуты накоротко, омическое сопротивление их невелико и, следовательно, не будь поляризационных сопротивлений, коррозия протекла бы весьма интенсивно. Всякое воздействие, уменьшающее поляризацию, увеличивает скорость коррозионных процессов. Факторы, уменьшающие поляризацию электродов коррозионного элемента, называют ДЕПОЛЯРИЗАТОРАМИ, а процессы, протекающие при этом – АНОДНОЙ И КАТОДНОЙ ДЕПОЛЯРИЗАЦИЕЙ. 3.4.1. Анодная поляризация Анодная поляризация обычно оказывает меньшее влияние на изменение разности потенциалов элемента по сравнению с катодной поляризацией. Анодный процесс, заключающийся в ионизации металла и гидратировании ионов по уравнению: Men+ + mH2O → Men+ ∙ mH2O, может протекать беспрепятственно только при условии беспрерывного отвода образующихся ионов из прианодной зоны. Торможение анодного процесса будет иметь место и в случае отставания скорости выхода ионов металла в раствор от скорости перетекания электронов на катодные участки. В результате анодной поляризации отрицательный заряд на металлической обкладке двойного слоя уменьшается, а следовательно, потенциал металла сдвинется в положительную сторону (или станет менее отрицательны). Таким образом, анодная поляризация происходит или в связи с тем, что скорость анодного процесса не поспевает за скоростью отвода электронов (перенапряжение ионизации металла), или потому, что из−за недостаточно быстрого отвода перешедших в раствор ионов металла повышается концентрация этих ионов в прианодной зоне (концентрационная поляризация). Чем легче протекает анодный процесс, тем меньше его поляризуемость. Следить за тем, легко ли протекает анодный процесс или он заметно тормозится, можно по ходу поляризационной анодной кри50 Потенциал, В вой. Эта кривая показывает зависимость потенциала электрода от плотности анодного тока на нем. Такая кривая показана на рис. 10. Удаление продуктов анодной реакции вызывает деполяризацию анода. Деполяризация анода может быть ускорена перемешиЕ Е ванием раствора, образованием о А таких продуктов коррозии, которые выпадают в осадок, а также путем образования комплексных соединений. Вследствие связывания в комплекс ионов металла, перешедших в раствор, концентрация их в прианодной зоне уменьшаПлотность тока, мА/см2 ется; анодная поляризация сниРис. 10. Анодная поляризаци- жается и переход ионов в расонная кривая твор, т.е. коррозия, усиливается. Например, медь и ее сплавы образуют в растворе аммиака хорошо растворимый комплексный ион 2+. Этим объясняется сильная коррозия меди и ее сплавов в аммиачных растворах. Поляризация анодного процесса в обычных условиях происходит в незначительной степени. Исключение представляет случай возникновения анодной пассивности. 3.4.2. Катодная поляризация Протекание катодной реакции могут тормозить два основных процесса: − торможение за счет трудности протекания самой реакции присоединения электрона деполяризатором, т. е. задержка в реакции nē + D (Dnē). Затруднение протекания катодного процесса по этой причине называется ПЕРЕНАПРЯЖЕНИЕМ РЕАКЦИИ КАТОДНОЙ ДЕПОЛЯРИЗАЦИИ; − торможение в процессе подвода к катодной поверхности деполяризатора (D) или отвода от катодной поверхности продуктов восстановления деполяризатора (Dnē). Затруднение катодного процесса по этой причине называется КОНЦЕНТРАЦИОННОЙ ПОЛЯРИЗАЦИЕЙ КАТОДА. 51 Всякий процесс захвата электрона на катоде, т.е. всякий процесс восстановления какого−либо вещества может являться катодной деполяризующей реакцией. Деполяризация катода может протекать различными путями: − ассимиляция электронов ионами: 2H+ +2ē H2, Fe3+ + ē Fe2+, Cu2+ + ē Cu+, S2O8– + ē 2SO42–; − ассимиляция электронов нейтральными молекулами: О2 + 4ē + 2Н2О 4ОН–, Cl2 + 2ē 2Cl–, Н2О2 + 2ē 2ОН–; − ассимиляция электронов нерастворимыми пленками Fe3O4 + 2ē + H2O 3FeO + 2OH–, Fe(OH)3 + ē Fe(OH)2 + OH–; − ассимиляция электронов органическими соединениями RO + 2ē + 4H+ RH2 + H2O, R + 2ē + 2H+ RH2, где R – радикал или органическая молекула. Наиболее часто встречаемыми и наиболее важными катодными деполяризационными процессами являются два: а) разряд и выделение водорода (деполяризатор ион водорода) 2H+ +2ē H2. В этом случае коррозионный процесс называется коррозией с ВОДОРОДНОЙ ДЕПОЛЯРИЗАЦИЕЙ. Этому виду коррозии подвержены электроотрицательные металлы в кислотах и весьма электроотрицательные металлы, например Mg, в воде и нейтральных растворах электролитов; б) восстановление атомов или молекул кислорода электронами, притекающими на катодных участках − КИСЛОРОДНАЯ ДЕПОЛЯРИЗАЦИЯ: О2 + Н2О + 4ē 4ОН–. Деполяризатором здесь является растворенный в электролите кислород. Этот вид коррозии является наиболее распространенным. Так корродирует большинство металлов в атмосфере, почве, нейтральных растворах электролитов. Иногда оба деполяризующие процесса протекают параллельно, как например, при коррозии железа в разбавленных растворах серной кислоты в присутствии кислорода воздуха. 52 3.4.3. Водородная деполяризация Протекание коррозии металла с водородной деполяризацией возможно, если (Me)обр < (н2)обр. (20) Согласно уравнению Нернста (н2)обр = (н2)ºобр + (RT2,3/F)lg(aH+/pH21/2), (21) о где (н2) обр – стандартный обратимый потенциал водородного электрода при всех температурах (при ан+ = 1 и рН2 = 1 ат); R – универсальная газовая постоянная. Ниже приведены значения обратимого потенциала водородного электрода (н2)обр при 25 оС и различных значениях рН среды и pН2. рн2, ат 5∙10 1 –7 0 + 0,186 0 рН среды 7 − 0,228 − 0,414 14 − 0,641 − 0,828 Изменение потенциала катода при катодной деполяризации выделением водорода подчиняется ближе всего логарифмической зависимости от плотности тока. Эта зависимость определяется выражением: К = Кº – (а + b lg i). (22) Для очень малых плотностей катодного тока (меньше 10–4 – 10–5 А/см2) потенциал от плотности тока имеет линейную зависимость. Поэтому при i = 0 К = Кº, а не бесконечности, как это следует из уравнения. Поляризуемость катода при выделении на нем водорода принято определять ВЕЛИЧИНОЙ ПЕРЕНАПРЯЖЕНИЯ ВОДОРОДА на данном материале катода. Под ПЕРЕНАПРЯЖЕНИЕМ ВОДОРОДА понимают сдвиг потенциала катода при данной плотности тока в отрицательную сторону по сравнению с потенциалом водорода в том же растворе (без наложения тока) и обозначают через η η = − (К − Кº) (23) или η = a + blgI, (24) где b – константа, не зависящая от материала, рассчитывается как 2,3 ∙ 2RT / nF; а − константа, зависит от природы и состояния поверхности материала катода и численно равна величине перенапряжения при плотности тока, равной единице. 53 Поляризационная кривая для случая водородной деполяризации имеет вид кривой Кº MN (рис. 11). − N 1 M η Рис. 11. Катодная поляризационная кривая для процесса водородной деполяризации ко или (н2)обр i1 0 i P Если по оси абсцисс взять не плотность тока, а логарифм этой величины, то кривая перенапряжения будет прямой. В этом случае константа b является тангенсом угла наклона полученной прямой к оси абсцисс, константа а равна величине ординаты lg i = 0 (т.е. при плотности тока i = 1). Величина перенапряжения водорода зависит от плотности тока, т.е. при данной силе тока, от истинных размеров поверхности катода. При одинаковой силе тока она больше на гладкой поверхности, чем на шероховатой. 3.4.4. Кислородная деполяризация Процессы коррозии с КИСЛОРОДНОЙ ДЕПОЛЯРИЗАЦИЕЙ могут протекать в том случае, если при данных условиях обратимый потенциал металла отрицательнее обратимого потенциала кислородного электрода (Me)обр < (О2)обр. (25) 54 Обратимый потенциал кислородного электрода может быть вычислен по уравнению Нернста: (О2)обр = (О2)ºобр + (RT2,3/4F)lg(pО2/a4ОH−) (26) где (О2)ºобр – стандартный обратимый потенциал кислородного электрода (при аOH– = 1 и рО2 = 1 ат); рО2 – парциальное давление кислорода; аОН– – активность гидроксильных ионов. Величина обратимого потенциала кислородного электрода (О2)ºобр при 25 oC для различных значений рН и рО2: рН среды ро2 , ат 0,21 1 0 + 1,218 + 1,229 7 + 0,805 + 0,815 14 + 0,391 + 0,400 Коррозия с кислородной деполяризацией является термодинамически более возможным процессом, чем коррозия с водородной деполяризацией, так как равновесный потенциал восстановления кислорода более положителен, чем равновесный потенциал выделения водорода. Коррозия металлов с кислородной деполяризацией является самым распространенным коррозионным процессом. Коррозии с кислородной деполяризацией подвергаются металлическая обшивка нефтезаводской аппаратуры и оборудования, детали аппаратов, соприкасающиеся с водой и нейтральными водными растворами солей (например, трубы конденсационно−холодильного оборудования), подземные трубопроводы, днища резервуаров и др. При коррозии металлов с кислородной деполяризацией катодный процесс определяется перенапряжением катодной реакции и скоростью подвода кислорода к электроду. Замедленность катодной реакции по схеме: О2 + 2Н2О + 4е 4ОН– (в щелочных или нейтральных растворах), или по схеме: О2 + 4Н+ + 4е 2Н2О (в слабо−кислых растворах) называется ПЕРЕНАПРЯЖЕНИЕМ ИОНИЗАЦИИ КИСЛОРОДА. При больших плотностях тока (при iк > 10–2 A/m2) i znacznej szybkości dopływu tlenu do katody, przepięcie jonizacji tlenu ma logarytmiczną zależność od gęstości prądu 55 δ" = а"+ b" log i, (27) gdzie δ" to przepięcie jonizacji tlenu; a” jest stałą zależną od materiału katody i stanu jej powierzchni, temperatury i innych czynników. Jest definiowana numerycznie jako wartość przepięcia przy i = 1; b” jest stałą niezależną od materiału katody; wyznacza się ją z równości b" = 2RT/nF ∙ 2,3; w temperaturze 20° i n = 1, b" = 0,117. Mechanizm katodowej redukcji tlenu jest dość złożony i przebiega w kilku etapach: rozpuszczanie tlenu w elektrolicie, przenoszenie rozpuszczonego tlenu do katodowych odcinków metalu, jonizacja tlenu. W przeciwieństwie do depolaryzacji wodoru, dla której polaryzacja stężeniowa jest praktycznie nieistotna, w korozji metali z depolaryzacją tlenową odgrywa ona bardzo ważną rolę ze względu na ograniczoną szybkość dostarczania tlenu do katody. Tłumaczy się to małą rozpuszczalnością tlenu w roztworze elektrolitu, a co za tym idzie jego niskim stężeniem, trudnością w dyfuzji tlenu przez nieruchomą warstwę cieczy sąsiadującą z katodą. Przy silnym mieszaniu roztworu, intensywnej cyrkulacji lub w warunkach zapewniających znaczne napowietrzenie elektrolitu, proces korozji jest hamowany przez nadmierne napięcie jonizacji tlenu. Procesy dyfuzyjne wpływają na szybkość korozji, gdy metal zanurzony jest w spokojnym lub lekko mieszanym roztworze. Ogólna krzywa polaryzacji katodowej ma postać zespoloną (rys. 12) i jest sumą trzech krzywych charakteryzujących polaryzację podczas jonizacji tlenu (AB), polaryzację stężeniową (DE) i wyładowanie jonów wodorowych (GFH). W pierwszej części omówiono prędkość procesu katodowego, tj. ilość tlenu zredukowana w jednostce czasu jest mniejsza niż maksymalna możliwa szybkość dostarczania tlenu na powierzchnię w drodze dyfuzji. Pod tym względem w tym obszarze prędkość procesu jest ograniczona głównie przez powolność samego procesu redukcji tlenu. 56 − H F K E G (H2)arr 0 (O2)arr A D B C iä i Rys. 12 Krzywa polaryzacji katody dla procesu depolaryzacji tlenu Wraz z dalszym wzrostem gęstości prądu potencjał przesuwa się w kierunku ujemnym, najpierw stopniowo, a potem nagle (sekcja CDE). Gwałtowne przesunięcie potencjału w kierunku ujemnym wiąże się z polaryzacją stężenia w wyniku spadku stężenia tlenu. Przy pewnej wartości potencjału możliwy staje się nowy proces katodowy, zwykle związany z depolaryzacją wodoru (sekcja EK). 3.5. Kontrolowanie czynnika korozyjnego Proces korozji elektrochemicznej metali i stopów składa się z procesów elementarnych lub etapów. Rzeczywista prędkość procesu korozji jest bezpośrednio zależna od jego całkowitego zahamowania na każdym etapie. Procent zahamowania całego procesu na każdym jego etapie charakteryzuje STOPIEŃ KONTROLI PROCESU na danym etapie. 57 nazywany jest etapem określającym szybkość korozji, ponieważ ma największą odporność w porównaniu z innymi etapami. PROCES KONTROLNY o2)arr imax I imax b b b a (o2)arr in A A imax (Me)arr (Me)arr K ∆K R P (o2)arr imax I I S (о2)arr i I g g d d Ryc. 13. Diagramy korozji polaryzacyjnej dla głównych praktycznych przypadków monitorowania procesów korozji elektrochemicznej 58 I w W warunkach praktycznych istnieje kilka podstawowych przypadków monitorowania korozji elektrochemicznej metali i stopów, uzasadnionych przez N.D. Tomaszow. Bekkerev I.V. „Metale i stopy - gatunki, skład chemiczny”, część 2, Uljanowsk: Państwowy Uniwersytet Techniczny w Uljanowsku, 2007. - s. 630, http://www.bibliotekar.ru/spravochnik-73/index.htm. 3]. Semenova I.V. Korozja i ochrona przed korozją, M.: Fizmatlit, 2002, 335 s. 4]. Khaldeev G.V., Borisova T.F. „Przepuszczalność wodoru przez metale i stopy w procesach korozyjno-elektrochemicznych. Elektrochemia” – tom 30, Moskwa, 1989 s. 232 5]. T. L. Lukanina, T. T. Ovchinnikova, V. Ya Sigaev, „Chemia ogólna” Część II. Instruktaż. wydanie 2, poprawione i rozszerzone, 2006)