Chemická odolnost stavebních materiálů v závislosti na jejich složení a struktuře. Technologické vlastnosti materiálů. Chemická odolnost materiálů Vladislav Aleksandrovich Likhachev x Hlavní průběh práce
Ministerstvo všeobecného a odborného školství
Ruská Federace
MOSKVA STÁTNÍ UNIVERZITA ENVIRONMENTÁLNÍHO INŽENÝRSTVÍ
A.G.Parshin V.S.Pakhomov D.L.Lebeděv
Chemická odolnost materiálů a ochrana proti korozi
Laboratorní dílna
Editoval Dr. Tech. vědy A.A
Moskva-1998
BBK35.11 Χ 46
Recenzenti:
Katedra koroze Moskevské státní akademie ropy a zemního plynu pojmenovaná po. Gubkin;
Ph.D. tech. Sciences A.S. Abramov, ekologická společnost "ShanEco", Moskva.
Schváleno jako učební pomůcka redakční a vydavatelskou radou Moskevské státní univerzity environmentálního inženýrství.
Parshin A.G., Pakhomov V.S., Lebedev D.L.
Χ 46 Chemická odolnost materiálů a ochrana proti korozi: Laboratorní dílna / Ed. A.A. Ševčenko - M.: MSUIE, 1998.-80 s.; nemocný.8.
ISBN 5-230-11142-9
Workshop obsahuje čtyři laboratorní práce o elektrochemické korozi a ochraně kovů: „Vliv strukturní heterogenity na korozi kovů při redukci vodíkových iontů“, „Koroze kovů s depolarizací kyslíku“, „Elektrodové potenciály“, „Kontaktní koroze a katodická ochrana kovů před korozí“. Jsou nastíněny základní pojmy teorie elektrochemické koroze kovů a metodika provádění výzkumu koroze. Určeno pro studenty 3. a 4. ročníku prezenční formy studia předmětu Chemická odolnost materiálů a ochrana proti korozi.
ISBN 5-230-11142-9 UDC620.193 BBK 35.11
© A.G.Parshin, V.S.Pakhomov, D.L.Lebedev.1998
© MSUIE, 1998
Předmluva
Laboratorní workshop je napsán v souladu s programem kurzu „Chemická odolnost materiálů a ochrana proti korozi“, který je součástí osnov řady specializací. Součástí workshopu byly práce na elektrochemické korozi kovů, která byla vyvinuta na Katedře kompozitních materiálů a protikorozní ochrany.
Každá laboratorní práce začíná teoretickou částí. Je zvažován mechanismus, termodynamika a kinetika koroze při katodické redukci vodíkových iontů, depolarizace kyslíku, rovnovážné a nerovnovážné procesy na rozhraní kov-elektrolyt a teorie elektrodových potenciálů.
Při prezentaci teoretických problémů jsou využívány moderní elektrochemické koncepty o mechanismu a kinetice korozních procesů.
Provádění laboratorních prací umožňuje studentům lépe porozumět základům studia koroze a ochrany kovů a také vštěpuje dovednosti při provádění základního laboratorního korozně-elektrochemického výzkumu.
Vedení laboratorního deníku
1) název a účel díla;
2) instalační schéma;
3) tabulka s výsledky experimentů a jejich zpracování (výpočty, grafy);
4. závěr.
Deník by měl být udržován úhledně a okamžitě čistý, tzn. stejným způsobem jako jsou vedeny záznamy o všech výzkumných a testovacích činnostech.
Schéma instalace by mělo být jasné a srozumitelné bez slovního vysvětlování.
Je nutné zajistit experimentální podmínky: testované materiály, povrch vzorků, složení a koncentraci elektrolytů, teplotu atd.
Výsledky zkoušek by měly být zaznamenány do předem sestavených tabulek, jejichž formy jsou uvedeny v popisu práce.
Při evidenci různých veličin je nutné uvést jejich rozměr. Výsledky měření zpravidla podléhají dalšímu zpracování - analytickému a grafickému (výpočet hmotnostních, objemových a hloubkových ukazatelů koroze, potenciálů atd.). V těchto případech je nutné uvést kalkulační vzorec a jeden výpočet v plném rozsahu, tzn. se nahrazením experimentálních hodnot do vzorce a pro další podobné výpočty - pouze konečné výsledky.
Na základě získaných výsledků a odpovídajícím způsobem zpracovaných je třeba vyvodit stručné závěry o provedené práci. Po dokončení odevzdejte žurnál s experimentálními údaji učiteli na vízum.
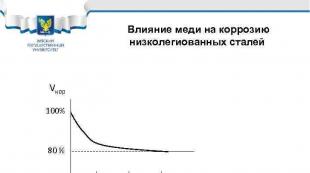
 Vliv mědi na korozi nízkolegovaných ocelí Vcor 100% 80% 0. 1 0. 2 0. 3% Cu Příklady ocelí: 10 HSND, 10 G 2 S 1 D, 10 KhDNP, 09 G 2 D, 18 G 2 AF(D)
Vliv mědi na korozi nízkolegovaných ocelí Vcor 100% 80% 0. 1 0. 2 0. 3% Cu Příklady ocelí: 10 HSND, 10 G 2 S 1 D, 10 KhDNP, 09 G 2 D, 18 G 2 AF(D)
 Klasifikace korozivzdorných ocelí 1. Korozivzdorné (nerezové) oceli a slitiny jsou materiály, které odolávají elektrochemické korozi v elektrolytech. 2. Hlavním legujícím prvkem korozivzdorného legování je chrom. 3. Chrom se do korozivzdorných ocelí zavádí v souladu s Tammannovým pravidlem. 4. Podle prostředí, ve kterém se tyto oceli používají, se rozlišuje pět skupin korozivzdorných (nerezových) ocelí a slitin.
Klasifikace korozivzdorných ocelí 1. Korozivzdorné (nerezové) oceli a slitiny jsou materiály, které odolávají elektrochemické korozi v elektrolytech. 2. Hlavním legujícím prvkem korozivzdorného legování je chrom. 3. Chrom se do korozivzdorných ocelí zavádí v souladu s Tammannovým pravidlem. 4. Podle prostředí, ve kterém se tyto oceli používají, se rozlišuje pět skupin korozivzdorných (nerezových) ocelí a slitin.
 Korozivzdorné oceli pro mírně agresivní prostředí Oceli první skupiny mohou pracovat pouze v uzavřené atmosféře a podvodní korozi s povinným periodickým sušením. V podmínkách otevřené atmosféry a stálé podvodní koroze (zejména v horké vodě), jakož i podzemní koroze, tyto oceli podléhají důlkové korozi. Tyto oceli zahrnují chromové oceli: 08 X 13, 09 X 13, 08 X 17 G (feritické), 10 X 13, 12 X 13 (martenziticko-feritické), 20 X 13, 30 X 13, 40 X 13 (martenzitické) . Stejně jako chrommanganové a chromniklové oceli s ekonomickým legováním niklem (2 -4%) 15 X 17 AG 14, 10 X 14 AG 15, 10 X 14 G 14 N 3 T, 12 X 17 G 14 N 3 , 08 X 18 G 8 N 2 T
Korozivzdorné oceli pro mírně agresivní prostředí Oceli první skupiny mohou pracovat pouze v uzavřené atmosféře a podvodní korozi s povinným periodickým sušením. V podmínkách otevřené atmosféry a stálé podvodní koroze (zejména v horké vodě), jakož i podzemní koroze, tyto oceli podléhají důlkové korozi. Tyto oceli zahrnují chromové oceli: 08 X 13, 09 X 13, 08 X 17 G (feritické), 10 X 13, 12 X 13 (martenziticko-feritické), 20 X 13, 30 X 13, 40 X 13 (martenzitické) . Stejně jako chrommanganové a chromniklové oceli s ekonomickým legováním niklem (2 -4%) 15 X 17 AG 14, 10 X 14 AG 15, 10 X 14 G 14 N 3 T, 12 X 17 G 14 N 3 , 08 X 18 G 8 N 2 T
 Korozivzdorné (nerezové) oceli pro slaná prostředí Druhá skupina korozivzdorných (nerezových) ocelí se používá ve slaném prostředí při nízkých teplotách, zejména při mořské korozi. Zvýšené korozní odolnosti je dosaženo dodatečným ekonomickým legováním Ni ocelí (5 – 8 %). Příklady: 09 X 15 N 8 Yu, 07 X 16 N 6, 08 X 17 N 5 M 3 (ocel se používá v prostředí s kyselinou sírovou), 09 X 17 N 7 Yu 1 (oceli se používají v podmínkách mořské koroze).
Korozivzdorné (nerezové) oceli pro slaná prostředí Druhá skupina korozivzdorných (nerezových) ocelí se používá ve slaném prostředí při nízkých teplotách, zejména při mořské korozi. Zvýšené korozní odolnosti je dosaženo dodatečným ekonomickým legováním Ni ocelí (5 – 8 %). Příklady: 09 X 15 N 8 Yu, 07 X 16 N 6, 08 X 17 N 5 M 3 (ocel se používá v prostředí s kyselinou sírovou), 09 X 17 N 7 Yu 1 (oceli se používají v podmínkách mořské koroze).
 Oceli pro použití v prostředích se střední korozí Prostředí se střední korozí znamenají roztoky solí při různých teplotách a také slabé roztoky některých kyselin. Oceli třetí skupiny jsou nejběžnější nerezové oceli s širokým uplatněním. Mezi tyto oceli rozlišujeme: a) oceli - náhrady za vysokoniklové oceli: 15 X 25 T, 15 X 28, 08 X 22 N 6 T, 12 X 21 N 5 T. b) oceli s optimálním obsahem chromu až niklu poměr (Cr: Ni = 18: 9, 18: 10): 12 X 18 N 9 T a 12 X 18 N 10 T, 17 X 18 N 9, 12 X 18 N 10 B, 08 X 18 N 10, 12 X 18 N 12 T, 08 X 18 N 12 B, 06 X 18 N 11 atd.
Oceli pro použití v prostředích se střední korozí Prostředí se střední korozí znamenají roztoky solí při různých teplotách a také slabé roztoky některých kyselin. Oceli třetí skupiny jsou nejběžnější nerezové oceli s širokým uplatněním. Mezi tyto oceli rozlišujeme: a) oceli - náhrady za vysokoniklové oceli: 15 X 25 T, 15 X 28, 08 X 22 N 6 T, 12 X 21 N 5 T. b) oceli s optimálním obsahem chromu až niklu poměr (Cr: Ni = 18: 9, 18: 10): 12 X 18 N 9 T a 12 X 18 N 10 T, 17 X 18 N 9, 12 X 18 N 10 B, 08 X 18 N 10, 12 X 18 N 12 T, 08 X 18 N 12 B, 06 X 18 N 11 atd.
 Oceli pro použití v prostředí se zvýšenou korozí Tyto typy ocelí byly vyvinuty pro zvýšení chemické odolnosti v horkých roztocích Na. Cl a v kyselých roztocích. Pro zvýšení odolnosti ocelí se používá dodatečné legování molybdenem a mědí a u ocelí této skupiny se často snaží zachovat austenitickou strukturu, což je výhodné z technologického hlediska, což vyžaduje dodatečné legování ocelí niklem. Vzhledem k vysokému obsahu legujících složek, především niklu, jsou oceli této skupiny poměrně drahé. Příklady ocelí ve skupině jsou: 10 X 17 N 13 M 2 T 08 X 17 N 13 M 3 T, 08 X 17 N 15 M 3 T, 04 X 28 MDT, 03 X 28 MDT, 06 X 28 MT.
Oceli pro použití v prostředí se zvýšenou korozí Tyto typy ocelí byly vyvinuty pro zvýšení chemické odolnosti v horkých roztocích Na. Cl a v kyselých roztocích. Pro zvýšení odolnosti ocelí se používá dodatečné legování molybdenem a mědí a u ocelí této skupiny se často snaží zachovat austenitickou strukturu, což je výhodné z technologického hlediska, což vyžaduje dodatečné legování ocelí niklem. Vzhledem k vysokému obsahu legujících složek, především niklu, jsou oceli této skupiny poměrně drahé. Příklady ocelí ve skupině jsou: 10 X 17 N 13 M 2 T 08 X 17 N 13 M 3 T, 08 X 17 N 15 M 3 T, 04 X 28 MDT, 03 X 28 MDT, 06 X 28 MT.
 Slitiny na bázi niklu pro velmi agresivní prostředí Médii s velmi vysokou agresivitou se rozumí horké roztoky kyseliny sírové a chlorovodíkové. V takto agresivním prostředí jsou nejodolnější kovové materiály slitiny na bázi niklu. Například slitina KhN 65 MV je stabilní při zvýšených teplotách v prostředí kyseliny sírové a chlorovodíkové, v koncentrované kyselině octové. Slitina N 70 MF se doporučuje pro použití v roztocích kyseliny sírové a chlorovodíkové, slitina je odolnější vůči mezikrystalové korozi.
Slitiny na bázi niklu pro velmi agresivní prostředí Médii s velmi vysokou agresivitou se rozumí horké roztoky kyseliny sírové a chlorovodíkové. V takto agresivním prostředí jsou nejodolnější kovové materiály slitiny na bázi niklu. Například slitina KhN 65 MV je stabilní při zvýšených teplotách v prostředí kyseliny sírové a chlorovodíkové, v koncentrované kyselině octové. Slitina N 70 MF se doporučuje pro použití v roztocích kyseliny sírové a chlorovodíkové, slitina je odolnější vůči mezikrystalové korozi.
 Zvýšení hustoty betonu 4. Zavedení polymerních přísad 4. 1. přidání malého množství 0,2 - 3 % polymerních přísad do betonové směsi (latexy, polymerní pryskyřice); 4. 2. výroba betonu na bázi polymerního pojiva (polymerní roztoky a polymerbeton); Dodává se jako suchá směs a tužidlo v plechovkách. 4. 3. impregnace hotových betonových a železobetonových výrobků polymerními sloučeninami nebo monomery s jejich následnou polymerací přímo v betonovém tělese (polymery betonu); 4. 4. armování betonu polymerními vlákny (výroba vláknobetonu)
Zvýšení hustoty betonu 4. Zavedení polymerních přísad 4. 1. přidání malého množství 0,2 - 3 % polymerních přísad do betonové směsi (latexy, polymerní pryskyřice); 4. 2. výroba betonu na bázi polymerního pojiva (polymerní roztoky a polymerbeton); Dodává se jako suchá směs a tužidlo v plechovkách. 4. 3. impregnace hotových betonových a železobetonových výrobků polymerními sloučeninami nebo monomery s jejich následnou polymerací přímo v betonovém tělese (polymery betonu); 4. 4. armování betonu polymerními vlákny (výroba vláknobetonu)

 Modul 7. Metody ochrany kovů před elektrochemickou korozí. Přednáška 7. 3 ELEKTROCHEMICKÁ OCHRANA KOVŮ PŘED KOROZI Katodická ochrana spočívá v posunutí potenciálu kovu korodující struktury na zápornou stranu připojením k zápornému pólu zdroje proudu.
Modul 7. Metody ochrany kovů před elektrochemickou korozí. Přednáška 7. 3 ELEKTROCHEMICKÁ OCHRANA KOVŮ PŘED KOROZI Katodická ochrana spočívá v posunutí potenciálu kovu korodující struktury na zápornou stranu připojením k zápornému pólu zdroje proudu.
 Modul 7. Metody ochrany kovů před elektrochemickou korozí. Přednáška 7. 3 Korozní schéma katodové ochrany
Modul 7. Metody ochrany kovů před elektrochemickou korozí. Přednáška 7. 3 Korozní schéma katodové ochrany
 Modul 7. Metody ochrany kovů před elektrochemickou korozí. Přednáška 7. 2 Ochranná ochrana je založena na charakteristikách koroze dvou kovů ve styku. Podle teorie kontaktní koroze se při kontaktu kladného kovu M 2 s negativnějším kovem M 1 posouvá potenciál kovu M 2 na zápornou stranu a jeho koroze klesá nebo se úplně zastaví.
Modul 7. Metody ochrany kovů před elektrochemickou korozí. Přednáška 7. 2 Ochranná ochrana je založena na charakteristikách koroze dvou kovů ve styku. Podle teorie kontaktní koroze se při kontaktu kladného kovu M 2 s negativnějším kovem M 1 posouvá potenciál kovu M 2 na zápornou stranu a jeho koroze klesá nebo se úplně zastaví.
 Modul 7. Metody ochrany kovů před elektrochemickou korozí. Přednáška 7. 3 Anodická ochrana se používá pouze pro kovy náchylné k pasivaci v korozním prostředí. Jde o přesun kovového potenciálu z oblasti aktivního rozpouštění do oblasti pasivace pomocí externího zdroje proudu.
Modul 7. Metody ochrany kovů před elektrochemickou korozí. Přednáška 7. 3 Anodická ochrana se používá pouze pro kovy náchylné k pasivaci v korozním prostředí. Jde o přesun kovového potenciálu z oblasti aktivního rozpouštění do oblasti pasivace pomocí externího zdroje proudu.
 Modul 7. Metody ochrany kovů před elektrochemickou korozí. Přednáška 7. 3 Korozní schéma anodické ochrany
Modul 7. Metody ochrany kovů před elektrochemickou korozí. Přednáška 7. 3 Korozní schéma anodické ochrany
MINISTERSTVO ŠKOLSTVÍ A VĚDY RUSKÉ FEDERACE FEDERÁLNÍ STÁTNÍ VZDĚLÁVACÍ INSTITUCE VYSOKÉHO ODBORNÉHO ŠKOLSTVÍ "ST. PETERBURG STÁTNÍ TECHNOLOGICKÁ UNIVERZITA ROSTLINNÝCH POLYMERŮ" T. L. Lukanina, I. Michajlova, A. M. M. M Učebnice St. −Petersburg 2014 1 UDC 546(075) BBK 24,1я7 L 840 Lukanina T. L., Mikhailova I. S., Radin M. A. Chemická odolnost materiálů a ochrana proti korozi: učebnice. manuál − Petrohrad: SPbGTURP, 2014. – 85 s. Příručka poskytuje moderní představy o termodynamice a kinetice chemické koroze, mechanismech a různých typech elektrochemické koroze s přihlédnutím k prostředím a podmínkám charakteristickým pro zařízení chemického průmyslu. Jsou nastíněny vědecké principy výroby korozivzdorných ocelí a slitin a způsoby ochrany konstrukčních materiálů před korozí. Učebnice je určena studentům technických vysokých škol a fakult v technologických oborech. Může být užitečný pro postgraduální studenty, vědecké a inženýrské pracovníky chemických podniků, projekční a výzkumné organizace zabývající se ochranou kovů před korozí. Recenzenti: Dr. Chem. věd, prof. Katedra chemie ITMO státu Petrohrad. fakulta Slobodov A. A.; Ph.D. chem. vědy, docent Katedra organické chemie, St. Petersburg State Technical University of Turkmenistan, Evdokimov A.N. Doporučeno k publikaci Univerzitní redakční a vydavatelskou radou jako učební pomůcka. Redaktor a korektor T. A. Smirnova Tech. Editor L.Ya Titova Templan 2014, pos. 45 ______________________________________________________________ Předloženo k uveřejnění 14.04.14 Formát 60×84/16. Typ papíru č. 1. Ofsetový tisk. Tištěno l.5.5. Uch.-ed. 5.5 Náklad 100 výtisků Vydání č. 45. Cena "C". Ivana Chernykh, 4 St. Petersburg State Technological University of Plant Polymers, 2014 Lukanina T. L., Mikhailova I. S., Radin M. A., 2014 2 ÚVOD KOROZE materiálů je spontánní destrukce v důsledku chemické nebo elektrochemické interakce s prostředím. Destrukce kovů a slitin v důsledku koroze se liší od jiných typů spontánní destrukce, jako je eroze nebo opotřebení, které jsou způsobeny mechanickou interakcí s tělem nebo prostředím. U většiny kovů a slitin je za provozních podmínek stabilnější oxidovaný (iontový) stav, do kterého přecházejí v důsledku koroze. Termín "koroze" pochází z latinského slova "corrodere", což znamená "korodovat". Ztráty korozí se obvykle dělí na přímé a nepřímé. PŘÍMÉ ZTRÁTY zahrnují: − náklady na kov, který se mění v korozní produkty, a také náklady na zkorodované zařízení, které selže před uplynutím doby odpisování stanovené technickými podmínkami. Navíc náklady na výrobu zařízení zpravidla výrazně převyšují náklady na kov, ze kterého je vyrobeno; − zvýšení přídavků na korozi, což vede nejen ke zvýšení spotřeby kovu, ale také ke zhoršení designu. Například u potrubí se stejným vnějším průměrem vede zvýšený přídavek kovu ke snížení užitečného průřezu a v důsledku toho ke snížení průchodnosti potrubí; − náklady na ochranná opatření ve formě kovových a nekovových povlaků, korozivzdorných materiálů, inhibitorů atd. NEPŘÍMÉ ZTRÁTY korozí jsou způsobeny následujícími důvody: − dodatečné náklady na neplánované opravy zařízení, které selhalo; − nedostatečná výroba v důsledku odstávek pro opravy zařízení poškozených korozí; − možnost nehod, výbuchů a požárů; − únik produktů v důsledku tvorby průchozích otvorů ve stěnách zařízení v důsledku koroze; 3 – zmenšení průřezu potrubí v důsledku usazování korozních produktů, což vyžaduje další spotřebu energie pro udržení požadovaného výkonu proudění tekutiny; − snížení kvality produktu v důsledku kontaminace korozními produkty. Věda o korozi a ochraně kovů studuje interakci kovů a slitin s korozním prostředím, stanovuje mechanismus této interakce a její obecné principy. Vědeckým základem pro studium koroze a ochrany kovů je nauka o kovech a fyzikální chemie. Konečným praktickým cílem této vědy je ochrana před korozním ničením kovů při jejich zpracování a provozu zařízení. Kapitola 1. OBECNÉ INFORMACE O KOROZI 1.1. Termodynamika a kinetika korozních procesů Primární příčinou koroze je termodynamická nestabilita kovů v různých prostředích za daných vnějších podmínek. Zásadní možnost samovolného vzniku korozního procesu je dána zákony termodynamiky. Výskyt jakéhokoli spontánního procesu je doprovázen poklesem volné energie. Možnost samovolného výskytu korozních procesů lze posoudit změnami izobaricko-izotermického potenciálu ΔG1. Spontánní proces oxidace kovu - koroze - je možný, pokud ∆G< 0, то есть знак ―минус‖ означает, что изобарно−изотермический потенциал убывает и свободная энергия системы уменьшается. Наоборот, если ∆G > 0, pak znaménko plus pro izobaricko-izotermický potenciál odpovídá nárůstu volné energie, což ukazuje na nemožnost spontánního výskytu této reakce. IZOBARICKO-IZOTERMÁLNÍ POTENCIÁL (ΔG) – Gibbsova energie, jeden z termodynamických potenciálů, rovna ΔG = ΔН - TΔS, kde ΔН, ΔS, Т - entalpie, entropie, termodynamická teplota systému. ΔG je úměrné počtu částic v systému. Označuje se jako jedna částice a nazývá se chemický potenciál. ΔG je vhodný pro popis procesů, ve kterých je možná výměna mezi hmotou a okolními tělesy. V izotermickém rovnovážném procesu probíhajícím při konstantním tlaku se jedná o celkovou práci systému mínus práci proti vnějším tlakovým silám. Rovná se poklesu izobaricko-izotermického potenciálu (tj. maximální užitečné práci). 1 4 Ačkoliv však termodynamika umožňuje určit, jak daleko je zkoumaný systém od rovnovážného stavu a je schopen spontánní reakce, nevypovídá to o rychlosti tohoto procesu. Rychlost koroze je dána její kinetikou. Charakteristickým rysem korozních procesů je jejich heterogenní charakter. To je způsobeno skutečností, že k destrukci kovu dochází na rozhraní mezi dvěma fázemi, které mají různé stavy agregace, například na rozhraní kov-kapalina, kov-plyn. Za takových podmínek je určujícím faktorem pro kinetiku chemické interakce buď rychlost chemické interakce, nebo dodávka reagencií z objemů kontaktních fází na rozhraní, kde dochází ke korozní interakci, a naopak odvod reakčních produktů. směr. V prvním případě je rychlost koroze určena kinetickou kontrolou, ve druhém - difuzní kontrolou. V některých případech je rychlost koroze určena smíšenou difuzně-kinetickou kontrolou. Pro proces v ustáleném stavu jsou rychlost chemické reakce a rychlost difúze stejné. Proces koroze se skládá z několika fází, které mohou probíhat postupně nebo paralelně. Celková rychlost korozního procesu v ustáleném stavu sestávajícího z několika po sobě jdoucích fází je určena rychlostí nejpomalejšího stádia. Pokud jednotlivé fáze procesu probíhají paralelně, je celková rychlost koroze určena nejrychlejší fází. Pokud tedy jedna z etap probíhá mnohem rychleji než druhá, lze druhou etapu zanedbat. 1.2. Klasifikace korozních procesů podle mechanismu vzniku Bez ohledu na obrovskou rozmanitost podmínek pro vznik korozních procesů je lze klasifikovat podle dvou hlavních charakteristik: - podle mechanismu procesu - podle charakteru destrukce korozí . Existuje CHEMICKÁ a ELEKTROCHEMICKÁ koroze. 5 se vyskytuje v suchých plynech (plynová koroze) a v kapalných neelektrolytech2 (koroze v neelektrolytech). Není-li prostředím elektrolyt (suché plyny nebo kapalné neelektrolyty), pak k výměně elektronů dochází přímo mezi redukujícím kovem (červená) a jakoukoliv látkou obsahující elektronegativní prvek schopný k sobě přitahovat elektrony - oxidační činidlo (ox ), například: Fe + Сl2 → FeCl2 (1) CHEMICKÁ KOROZE červená Bilance přenosu elektronů ox Feº − 2ē → Fe2+ 1 Сl2º + 2ē → 2Cl− Feº + 2Сlº = Fe2+ + 2Cl− ELEKTROCHEMICKÝ proces interakce kovu s korozivním prostředím (roztok elektrolytu), ve kterém Ionizace atomů kovu a redukce oxidační složky korozivního prostředí neprobíhají v jednom aktu a jejich rychlosti závisí na potenciálu elektrody. Charakteristické rysy procesu elektrochemické koroze jsou následující: – současný výskyt dvou samostatných procesů - oxidačního (rozpouštění kovu) a redukce (vývoj vodíku, redukce kyslíku, uvolňování kovu z roztoku atd.); – proces rozpouštění kovu je doprovázen směrovým pohybem elektronů v kovu a iontů v elektrolytu, tzn. výskyt elektrického proudu; – produkty koroze vznikají v důsledku sekundárních reakcí. Redoxní procesy probíhající při elektrochemické korozi lze prezentovat ve formě následujících reakcí: Ме − nē→ Меn+ (anodický proces) (2) n− R(ox) + nē → R (červený) (katodový proces), (3 ) kde R(ox) je oxidační činidlo; NEELEKTROLYTY jsou látky, jejichž vodné roztoky a taveniny nevedou elektrický proud. Obsahují kovalentní nepolární nebo nízkopolární vazby, které se nerozpadají na ionty. Plyny, pevné látky (nekovy) a organické sloučeniny (sacharóza, benzín, alkohol) nevedou elektrický proud. 2 6 R n−(červená) – redukovaná forma oxidačního činidla; nē – počet přenesených elektronů. Příkladem elektrochemické koroze je proces oxidace (rezivění) železa vlivem vody: Fe − 2ē → Fe2+ (anodický proces – rozpouštění železa) H2O + ½О2 +2ē → 2OH– (katodový proces – redukce kyslíku) −−−−−− −−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−− − Fe 2+ + 2OH – → Fe(OH)2 (tvorba korozních produktů) Reakce (2) a (3) probíhají konjugovaně, ale řídí se vlastními kinetickými zákony. V tomto případě je nutné dodržet podmínky stacionárnosti procesu, tzn. rovnost rychlostí oxidace kovu a redukce oxidačního činidla. Tyto reakce mohou být geograficky odděleny a probíhají na různých částech povrchu. Z podmínek stacionarity vyplývá, že stačí zpomalit jednu z konjugovaných reakcí, aby se snížila rychlost celého procesu. 1.3. Klasifikace korozních procesů podle charakteru korozní destrukce Na základě charakteru korozní destrukce kovu se rozlišuje KOROZE OBECNÁ A MÍSTNÍ. OBECNÁ KOROZE se šíří po celém povrchu kovu; 1 2 4 5 7 8 3 6 9 Obr. 1. Typy korozního poškození Mnoho případů koroze zahrnuje obecnou korozi, například když v důsledku vystavení kovu elektrolytům nezůstávají na povrchu kovu korozní produkty. Intenzivní obecná koroze je tedy pozorována při interakci mědi s kyselinou dusičnou, 7 železa s kyselinou chlorovodíkovou, hliníku s žíravými alkáliemi, zinku s kyselinou sírovou atd. Jednotná obecná koroze je jedním z nejméně nebezpečných typů koroze za předpokladu, že rychlost rozpouštění kovu v důsledku poškození korozí nepřekračuje normy stanovené požadavky na provoz přístroje, zařízení apod. Pokud je kov dostatečně silný, koroze má malý vliv na snížení mechanické pevnosti konstrukce při rovnoměrně rozložených napětích po průřezu (tah, tlak). Obecná koroze však může být nebezpečná, když jsou díly vystaveny ohybu a kroucení, protože v tomto případě jsou zničeny nejvíce zatížené vrstvy. MÍSTNÍ KOROZE - způsobuje destrukci pouze některých oblastí kovového povrchu a zbytek nepodléhá destrukci nebo je míra destrukce velmi nízká. Projevy lokální koroze jsou velmi různorodé a mohou mít různou míru nerovností, a proto se lokální koroze dělí na KOROZE SKVRNAMI, VŘEDY, HRNCE (DOLKOVINY), SELEKTIVNÍ KOROZE, KOROZE, KOROZNÍ PRASKÁNÍ. INTERKRYSTALITOVÁ Koroze skvrnami, vředy, body - typy lokální koroze se liší poměrem průměru destruované plochy k její hloubce (obr. 1., 4–6). U bodové koroze je průměr zničené oblasti mnohem větší než její hloubka, u ulcerózní destrukce je průměr zničené oblasti úměrný její hloubce a u důlkové koroze je průměr zničené oblasti mnohem menší než její hloubka. . V některých případech může důlková koroze vést ke zničení, což je zvláště nebezpečné pro potrubí, nádrže atd. Důlková koroze je typická pro kovy schopné pasivace - brzdění (chrom, hliník, nerezové oceli atd.). Pasivní stav kovů může být narušen v určitých slabších oblastech povrchu, například v pórech ochranného filmu, v místech intermetalických vměstků (oxidy, strusky apod.). SELEKTIVNÍ KOROZE – dělí se na: komponentně selektivní (selektivní) nebo strukturně selektivní (interkrystalická). 8 Příkladem SLOŽKOVě SELEKTIVNÍ KOROZE je odzinkování mosazi. Odzinkování mosazi3 spočívá v tom, že zinek přechází do elektrolytu (obvykle při neutrální nebo mírně kyselé hodnotě pH) intenzivněji než měď. Na povrchu mosazi se tvoří uvolněná vrstva mědi, která přispívá ke zvýšené elektrochemické korozi (vzhled mikrogalvanického článku). Příkladem STRUKTURALNĚ-SELEKTIVNÍ koroze je koroze šedé litiny. Strukturní selektivní koroze šedé litiny spočívá v převládající destrukci feritové složky, jejímž výsledkem je vytvoření grafitového skeletu vyplněného korozními produkty (obr. 1−3). Současně prudce klesá mechanická pevnost litiny. INTERKRYSTALITOVÁ KOROZE je charakterizována selektivní destrukcí kovu podél hranic zrn (obr. 1–7). V důsledku toho může při malé změně hmotnosti a vzhledu dojít k velmi výrazné ztrátě pevnostních vlastností kovu (obr. 2). 40 Obr. 2. Ztráta pevnostních vlastností kovu při různých typech korozní destrukce 1 – rovnoměrná koroze; 2 – mezikrystalová koroze 20 Ztráta w, % 60 2 0 1 2 4 Ztráta hmotnosti, kg/cm2 V extrémních případech při úplném zničení hranic zrn v důsledku mezikrystalové koroze se slitina může rozpadat na prášek. KOROZNÍ PRASKÁNÍ je případ lokální koroze, při které dochází k destrukci ve směru kolmém na největší tahové napětí (obr. 1 - 8). 3 Mosaz – slitina mědi a zinku. 9 Typické je, že v tomto případě se korozní trhlina může šířit podél hranic zrn nebo rozříznout tělo krystalu. Destrukce korozní únavou (při současném působení korozního prostředí a střídavého nebo cyklického zatížení) probíhá obdobně jako při korozním praskání. PODPOVRCHOVÁ KOROZE, vycházející z povrchu, se šíří převážně pod povrchem, což často vede k delaminaci a bobtnání kovu (obr. 1− 9), např. k tvorbě bublin na nekvalitním plechu při procesu leptání nebo korozi při provozu nádob a přístrojů. 1.4. Indikátory koroze Pro kvantifikaci rychlosti koroze se používají indikátory koroze: MASOMETRICKÉ, OBJEMOVÉ, HLOUBKOVÉ, MECHANICKÉ, PROUDOVÉ. 2 MASOMETRICKÝ korozní index (Km, g/m ∙h) je změna hmotnosti kovu v důsledku koroze na jednotku povrchu (povrchů) vzorku a za jednotku času. Změna hmotnosti vzorku se obvykle určuje jako rozdíl mezi hmotností vzorku před testováním (m®) a jeho hmotností po testování (m1) v čase (τ) s odstraněním korozních produktů (ztráta hmoty kovu) . K m = (m0 − m1)/s ∙ τ . (4) 3 2 OBJEMOVÝ index koroze (Kν, cm /m h) je objem (Vo) plynu absorbovaného nebo uvolněného během procesu koroze na jednotku povrchu kovu a za jednotku času K m = V0 /s ∙ τ. (5) Index koroze HLOUBKY (P, mm/rok) zohledňuje zmenšení tloušťky kovu v důsledku koroze, vyjádřené v lineárních jednotkách a za jednotku času. Tento indikátor je užitečný při porovnávání koroze kovů s různou hustotou. Přechod od hmotnostního ukazatele k hloubkovému lze v případě rovnoměrné koroze provést pomocí vzorce P = K m ∙ 8,76 /γ, (6) kde γ je hustota kovu. Ukazatel MECHANICKÉ koroze je určen změnou jednoho z hlavních ukazatelů mechanických vlastností kovu za určitou dobu korozního procesu a vyjadřuje se v relativních jednotkách nebo v procentech. Pokud je pevnost v tahu použita jako mechanický indikátor, pak je změna indikátoru pevnosti (Kσ) určena vzorcem Kσ = Δσb/σb, (7) kde σb je pevnost v tahu před korozí, ∆σb je změna pevnosti v tahu kovu po korozi během času τ. Aktuální ukazatel koroze umožňuje pomocí Faradayova vzorce vypočítat množství zkorodovaného kovu na základě velikosti korozního proudu Km = IA /nF, (8) kde Km je rychlost koroze založená na změně v kovové hmotě; I – síla proudu; F – Faradayova konstanta; n – valence kovu; A je atomová hmotnost kovu. Při kvalitativním a kvantitativním hodnocení korozní odolnosti kovů se doporučuje používat desetibodovou stupnici4. Je třeba poznamenat, že i když se tato stupnice pro hodnocení korozní odolnosti kovů rozšířila zejména v chemickém průmyslu, není univerzální, neboť pro zařízení pro různé účely se přípustné rychlosti koroze mohou lišit o řád, resp. více. Tabulka 1 Desatero stupnice korozní odolnosti kovů a slitin Skupina odolnosti zcela perzistentní, velmi perzistentní odolná nízká odolná nestabilní rychlost koroze, mm/rok méně než 0,001 nad 0,001 do 0,005 do 0,01 do 0,01 do 0,05 nad 0,05 více 05. do 0,1 nad 0,1 do 0,5 nad 0,5 do 1,0 nad 1,0 do 5,0 nad 5,0 do 10,0 nad 10,0 SKÓRE 1 2 3 4 5 6 7 8 9 10 Jednotný systém ochrany proti korozi a stárnutí. Podzemní stavby. Všeobecné požadavky na ochranu proti korozi. GOST 9.602−2005, http://www.gosthelp.ru/gost/gost206.html 4 11 Kapitola 2. CHEMICKÁ KOROZE KOVŮ Chemická koroze kovů je spontánní proces jejich destrukce v důsledku chemické interakce s vnějším prostředím. Charakteristickými rysy procesu chemické koroze jsou: – realizace oxidačních a redukčních procesů v jedné fázi; – nepřítomnost elektrického proudu; – tvorba korozních produktů přímo na povrchu, kde dochází k jeho destrukci. CHEMICKÁ KOROZE je heterogenní reakce interakce mezi kapalnou (l) nebo plynnou fází (g) s pevnou fází (s) - kovem a dělí se na 2 typy (plynová koroze, koroze v kapalných neelektrolytech). 2.1. Plynová koroze V praxi má největší význam KOROZE PLYNU. Nejčastěji se projevuje jako koroze kovových materiálů za vysokých teplot (desetikrát vyšších než je bod varu vody) v atmosféře plynů obsahujících kyslík. Z tohoto důvodu se nazývá VYSOKOTEPLOTNÍ KOROZE nebo VYSOKOTEPLOTNÍ OXIDACE. Na povrchu výrobků vyrobených z kovů a slitin se v důsledku jejich interakce s takovým korozivním prostředím vytváří film výrobků představujících různé oxidy kovů. Současně mohou v závislosti na provozních podmínkách vznikat další sloučeniny, například sirné kovy5. Například v průmyslu rafinace ropy působí koroze plynu například na potrubí pecí, stěny reaktoru, reakční kolony a další zařízení vystavená působení sirovodíku, oxidu uhelnatého, dusíku, kyslíku a dalších plynů při vysokých teplotách. Pojem "plynová koroze" označuje destrukci kovů způsobenou působením par a plynů za vysokých teplot, i když je možné jej aplikovat i na nižší teploty při 5 SÍRNÉ KOVY, stejně jako sulfidy. 12 zajistit, aby na kovu nekondenzoval film kapaliny, který vede elektrický proud. Na povrchu výrobků vyrobených z kovů a slitin se v důsledku jejich interakce s korozivním prostředím vytváří film z výrobků představujících různé sloučeniny kovů. Obecně lze proces chemické koroze popsat např. reakcí oxidace kovu se zahřátým (T >>373K) plynem (G2): 2Ме + nГ2 → 2Men+Гn. V kyslíkovém prostředí lze takový proces popsat následující rovnicí Me(T) + ½ O2 ↔ MeO(T). Tato reakce je rovnovážná při vysokých teplotách. Opačný proces se nazývá OXIDOVÁ DISOCIACE. Kyslík přítomný v plynném prostředí je charakterizován svým parciálním tlakem (pO2). Chemická reakce oxidace kovu bude v rovnováze s reakcí disociace oxidu, pokud se parciální tlak kyslíku (pO2) a ELASTICITA DISOCIACE OXIDU (pMeO)7 vyrovnají (pMeO = pO2) 1. Když pO2 > pMeO, reakce pokračuje hlavně ve směru tvorby oxidů. Když pMeO > pO2, reakce probíhá převážně v opačném směru. Oxidace kovů je vícestupňový heterogenní proces, díky kterému se na povrchu vytvoří nejprve mono- a poté polymolekulární oxidová vrstva, tzv. oxidový film. Růst jeho tloušťky může probíhat buď v obou směrech současně (kovové i plynné prostředí), nebo převážně v jednom (obr. 3). ČÁSTEČNÝ TLAK (z pozdně latinské participace - částečný), tlak, který by měl plyn obsažený ve směsi plynů, kdyby sám zabíral objem rovný objemu směsi při stejné teplotě. 7 Rovnovážný parciální tlak kyslíku pro reakce vzniku nebo TEPELNÉ DISOCIACE OXIDŮ se nazývá ELASTICITA DISOCIACE OXIDŮ. Jeho hodnota souvisí s rovnovážnou konstantou reakce a je tedy funkcí teploty. 6 13 a b c Obr. Obr. 3. Schéma iontově-elektronického mechanismu tvorby a růstu oxidového filmu na povrchu kovového dílu Pokud k růstu dochází ve směru plynného prostředí, pak je pozorován výrazný nárůst velikosti dílu , například při oxidaci, se kterou je třeba ve strojírenství počítat. Směr růstu je určen poměrem mezi rychlostmi protidifúze kovových iontů Мen+ a kyslíku O2− uvnitř oxidového filmu. Pokud se rychlosti difúze kovových iontů (rdif.Me) a kyslíku (rdif.O2) velmi liší, pak k růstu oxidového filmu dochází převážně v jednom směru, například s rdiff.O2< rдиф.Mе − в направлении газовой среды и наоборот (рис. 3, в, б). В большинстве случаев, скорости диффузии ионов металла и кислорода соизмеримы, но для ионов кислорода скорость несколько ниже из−за того, что они больше по размерам, поэтому зона роста, находящаяся в толще самой оксидной пленки, располагается ближе к ее внешней поверхности (рис. 3, а). По мере утолщения оксидной пленки процессы встречной диффузии ионов и электронов затрудняются. С повышением температуры упругость диссоциации оксидов возрастает у всех металлов. Поэтому, начиная от какой−то температуры, когда упругость диссоциации оксида металла становится больше парциального давления кислорода в воздухе (при атмосферном давлении рО2 = 0,21 ат), металл перестает окисляться и становится вполне малоактивным по отношению к кислороду. Так, например, серебро при температуре 300 ºК термодинамически еще не устойчиво, а при 400 ºК и при всех более высоких температурах упругость диссоциации Ag2O превышает парциаль14 ное давление кислорода в воздухе. При температуре 2000 ºК медь тоже становится неокисляемым металлом, однако для таких металлов, как Fе, Zn, Ni и др., даже при этих температурах упругость диссоциации окислов остается еще достаточно низкой и, следовательно, протекание реакции окисления вероятно. Необходимо учесть, что температурная зависимость для кинетики процесса окисления совершенно иная, чем для термодинамической вероятности окисления. Поэтому нет никакого противоречия в том, что термодинамическая вероятность окисления металла с повышением температуры падает, а реальная скорость окисления (при условии, что термодинамически этот процесс остается еще возможным) с повышением температуры возрастает. 2.2. Образование тонких пленок на металлах Ранее было отмечено, что в большинстве случаев образовавшиеся продукты газовой коррозии – оксиды металла остаются на поверхности металла в виде пленки. Эти пленки определяют кинетику процесса и в случае наличия защитных свойств могут привести к замедлению коррозии. Чтобы пленка оксида металла обладала защитными свойствами, она должна быть: − сплошной; − прочно сцепляться с основным металлом и обладать близким к нему коэффициентом теплового расширения; − быть химически инертной по отношению к данной агрессивной среде; − обладать твердостью и износостойкостью. Даже при обычной температуре на поверхности многих металлов и сплавов на воздухе образуются слои оксидов. Еще в 1836 г. М. Фарадей высказал предположение, что даже на блестящей поверхности металлов существует тончайшая, невидимая защитная (оксидная) пленка. Этим он объяснял пассивность железа в азотной кислоте. Если пленка пористая, рыхлая и характеризуется плохим сцеплением с основным металлом, то даже при условии инертности в данной агрессивной среде, она не будет обладать защитными свойствами. Пленки по толщине (δ) делятся на три группы: − тонкие (невидимые), δ < 4000 нм; − средние (дающие цвета побежалости), δ = от 4000 до 50000 нм; 15 − толстые (видимые), δ > 50 000 nm. Tloušťka filmů vzniklých při interakci kovu se suchým vzduchem je různá a závisí na druhu kovu, teplotě a dalších faktorech. Tloušťka filmů na mědi a železe při pokojové teplotě je tedy 100–300 nm a na hliníku 1000–1500 nm. Uveďme několik příkladů, které dokazují přítomnost tenkých vrstev na kovech. 1. Když se čistý lesklý povrch železného plátu na jednom konci zahřeje, objeví se na něm interferenční barvy - „zakalené barvy“. To ukazuje na výskyt oxidů různé tloušťky. Barvy jdou v tomto pořadí - od studeného konce talíře k horkému: žlutá, oranžová, červená, fialová, fialová, modrá. Při testování ochranných vlastností vytvořeného filmu pomocí kapek roztoku dusičnanu měďnatého v místech, kde má film slabé ochranné vlastnosti, reaguje se železem: Cu(NO3)2 + Fe → Fe(NO3)2 + Cu. V důsledku toho se na povrchu pod kapkami objeví měď. Podle doby, po které dojde k reakci, lze posoudit ochranné vlastnosti filmů v různých oblastech. V tomto experimentu byly nejlepší ochranné vlastnosti zjištěny v oblasti, která se nachází mimo topné místo (za žlutou zónou), kde povrch zůstává nezměněn. Je zřejmé, že v této oblasti je velmi tenký, ale ve srovnání s vyhřívanými oblastmi hustší film. 2. Pokud zlomíte jehlu (nebo žiletku) pod rtutí, na zlomených místech se vytvoří amalgám. Když jsou rozbité vzorky umístěny ve vzduchu ve rtuti, není pozorováno sloučení. To se vysvětluje vytvořením velmi tenkého oxidového filmu, který zabraňuje amalgamaci. 3. Když se železo vloží do roztoku hydroxidu draselného s přebytkem jódu, udělá se na povrchu železa v roztoku hluboký vryp. Po nějaké době se železo v místě škrábnutí začne rozpouštět a na dně skla zůstávají vločky, které se po dalším zkoumání ukáží jako oxid železitý. Proces tvorby filmu na kovu probíhá ve fázích. 16 První fází procesu interakce kovu s korozním prostředím je chemisorpce oxidačního činidla (O2, CO2, H2O, SO2) na povrchu kovu, např.: Me(sol) + (O2) = Me( sol) + 2Oads. Za přítomnosti chemické afinity mezi kovem a oxidačním činidlem se realizuje druhý stupeň interakce kovu s korozním prostředím - přechod chemisorbovaného filmu na oxidový. Tento proces lze konvenčně popsat reakcí ve tvaru: xMe(s) + yOads = MexOy. Oxidový film vytvořený na povrchu kovu může zpomalit proces koroze v důsledku inhibice přívodu oxidačního činidla na povrch oxidačního kovu. V tomto případě MÁ fólie OCHRANNÉ VLASTNOSTI. 2.2.1. Podmínka pro spojitost filmů na kovech Ochranné vlastnosti mohou mít pouze SOUVISLÉ FÓLIE. Možnost vytvoření takového filmu je určena PODMÍNKOU KONTINUITY Pilling–Bedworth. Molární objem sloučeniny objevující se na povrchu kovu (Vok) musí být větší než objem kovu (VMe) spotřebovaný k vytvoření jednoho molu sloučeniny: Vok/VMe >1. Pokud Vok/VMe< 1, то пленка не может быть сплошной. К металлам, не удовлетворяющим условию сплошности при окислении их кислородом, относятся щелочные и щелочно−земельные металлы. Сплошность пленок – необходимый, но недостаточный фактор, определяющий ее защитные свойства. Как показал Францевич И. Н., у пленок с Vok/VMe >>1 nemohou mít vysoké ochranné vlastnosti kvůli výskytu velkých vnitřních pnutí v nich (například MoO3 nebo WO3). Za horní hranici objemového poměru se považuje 2,5. Pak zpřesněná podmínka spojitosti vypadá takto: 1< Vok/VMe < 2,5. 2.2.2. Законы роста оксидных пленок на металлах Процесс роста ПОРИСТОЙ ПЛЕНКИ не осложняется процессом подвода окислителя к поверхности металла, а контролируется скоростью химической реакции образования оксида. Скорость коррозии в этом случае выражается уравнением: 17 V = dx/d = kC, где x − толщина пленки; – время; k − константа скорости химической реакции образования оксида; C − концентрация окислителя на поверхности метала. Рост пористой пленки, контролируемый скоростью химической реакции окисления металла, протекает во времени ПО ЛИНЕЙНОМУ ЗАКОНУ. Линейный закон роста имеет место: − при высокотемпературном окислении на воздухе и в среде кислорода при Vok/VMe<1; − в случае образовании летучих оксидов. Сплошные пленки затрудняют проникновение реагентов друг к другу и их рост сопровождается самоторможением процесса. Результирующая скорость этого сложного процесса определяется скоростью самой медленной стадии, т.е. возможны различные варианты контроля течения процесса. 1−й вариант. Скорость коррозии контролируется стадией массопереноса (диффузионный контроль процесса). В этом случае концентрация окислителя на границе раздела Ме – оксидная пленка равна нулю, так как скорость химической реакции весьма велика. Уравнение скорости течения коррозии определяется ПАРАБОЛИЧЕСКИМ ЗАКОНОМ роста оксидной пленки. Параболический закон роста реализуется, в частности, при окислении железа на воздухе при различных температурах. 2−ой вариант. В предыдущем случае С = 0, т.е. накопление окислителя на внутренней поверхности оксидной пленки не происходит. Если учесть возможность кинетического торможения процесса роста оксидной пленки наряду с торможением на стадии массопереноса (диффузионно−кинетический контроль процесса), в этом случае приходим к СЛОЖНО − ПАРАБОЛИЧЕСКОМУ ЗАКОНУ роста оксидной пленки. Рост оксидных пленок по сложно−параболическому закону выполняется: а) при окислении железа в парах воды или в атмосфере СО2; б) при окислении Сo, Сu в атмосфере чистого кислорода; в) при частичном разрушении оксидной пленки. Рост оксидных пленок при диффузионно−кинетическом контроле может быть выражен ТАКЖЕ СТЕПЕННЫМ ЗАКОНОМ. 18 3−й вариант. Часто рост пленки протекает медленнее, чем это следует из параболического закона роста. Это наблюдается при возникновении оксидных пленок при низких температурах (на Сu в О2 при t < 100 оС; на Аl, Тi, Ni, Zn в О2 при t = 300400 оС.) Затухание объясняется либо уплотнением пленок, либо появлением дефектов (пузырей, расслоений). В этих случаях рост толщины пленки протекает в соответствии с логарифмическим законом роста: x = ln(K). 2.3. Термодинамика процесса химической коррозии В качестве примера рассмотрим процесс окисления металла в атмосфере кислорода. Процесс окисления металла может быть представлен реакцией вида: 2Ме + nО2 → Me2n+Оn. Движущая сила процесса коррозии (ДСП) может быть определена с помощью термодинамики. Торможение процесса (ТП) не может быть определено термодинамически, однако с помощью термодинамики можно оценить условия, уменьшающие или исключающие протекание процесса (применение защитных сред, катодная защита, обескислороживание и др.). Независимо от механизма коррозии возможность ее протекания определяется знаком изменения термодинамического потенциала (см. раздел 1.1). Любое изменение энергии системы характеризует переход ее в новое состояние и является мерой стабильности системы: ΔG = GII − GI , где ΔG – энергия, израсходованная на изменение состояния системы, например, на процесс коррозии; GI − энергия системы в исходном состоянии, например, металла в конкретных условиях; G II − энергия системы в новом состоянии, например, прокорродировавшего металла. Рассчитать ΔG можно, вспомнив начало химии8. Величину изобарно-изотермического потенциала можно рассчитать по разности между суммами Δ G продуктов реакции и исходных веществ: GХР = ΔG ni – ΔG mi. (прод. р-ции) (исх. в-в). ΔG - является функцией температуры. Стандартные значения для большинства веществ сведены в справочные таблицы термодинамических величин. Изменение энергии Гиббса в зависимости от температуры учитывается уравнением Гиббса. ΔG Т = ΔН 298 – 298 Δ S 298. ΔG простого вещества = 0. Определить энергию Гиббса для любой химической реакции можно по уравнению ΔG = ΔG + RТ ln К, рассчитав предварительно величину константы равновесия, например, по равновесным концентрациям веществ. При равновесии ΔG=0, поэтому ΔG+RТlnК=0, следова8 19 2.4. Кинетика процесса химической коррозии Хотя термодинамика и дает ответ на вопрос о том, насколько изучаемая система отдалена от состояния равновесия, но ответ на весьма важный практический вопрос о скорости протекания коррозионного разрушения в рамках термодинамики получен быть не может. Рассмотрением этого вопроса и занимается кинетика коррозионных процессов. 2.4.1. Скорость процесса коррозии. Показатели химической коррозии СКОРОСТЬ КОРРОЗИОННОГО процесса может быть представлена в общем виде с помощью уравнения: СКОРОСТЬ КОРРОЗИИ (СК) = ДСП / ТП, где ДСП − движущая сила процесса; ТП − торможение процесса. Скорость химической коррозии определяют количественно по наблюдениям во времени за изменением какой−либо подходящей для этих целей величины, изменяющейся в процессе коррозии. Тогда истинная скорость коррозии металлического материала определяется из выражения: V = dy/d, где y − изменяющаяся в процессе коррозии характеристика свойств материала; − время коррозии. Для количественного выражения скорости коррозионных разрушений на практике пользуются показателями коррозии, являющимися, по сути, отражением СРЕДНЕЙ СКОРОСТИ КОРРОЗИОННОГО разрушения материала: Vср=∆y/∆. Показатели коррозии. 1. ГЛУБИННЫЙ ПОКАЗАТЕЛЬ коррозии: Kn=∆П/∆, где ∆П − глубина (средняя или максимальная) коррозионного разрушения; ∆ − промежуток времени коррозии. 2. ПОКАЗАТЕЛЬ ИЗМЕНЕНИЯ ТОЛЩИНЫ образующейся на металле пленки продуктов коррозии: Kn= ∆h/∆, где ∆h − изменение толщины образующейся на металле пленки продуктов коррозии. тельно, ΔG = – RТlnК. Данное уравнение позволяет также определить значение константы равновесия химической реакции по величине изобарно-изотермического потенциала. Для определения направления протекания многих химических реакций достаточно оценить знак Δ G. 1). При ΔН 0 , Δ S 0 реакция самопроизвольна (Δ G 0) при Т. 2). При ΔН 0, Δ S 0 реакция обратная (Δ G 0) при любой Т. 3). При ΔН 0, Δ S 0, реакция самопроизвольна (Δ G 0) при любой Т. 4). При ΔН 0, ΔS 0 реакция самопроизвольна (Δ G 0) при Т. 20 3. ПОКАЗАТЕЛЬ ИЗМЕНЕНИЯ МАССЫ Km=∆m/S∆, где ∆m − изменение массы корродирующего металла; S – площадь поверхности коррозии. 4.ОБЪЕМНЫЙ ПОКАЗАТЕЛЬ КОРРОЗИИ Kv=∆V/S∆, где ∆V − объем газа, выделившегося или поглощенного в процессе коррозии и приведенный к нормальным условиям. 5. МЕХАНИЧЕСКИЙ ПОКАЗАТЕЛЬ КОРРОЗИИ Кs=(∆s/∆)100 %, где ∆s − относительное изменение характеристики механического свойства. 6. ПОКАЗАТЕЛЬ ИЗМЕНЕНИЯ ЭЛЕКТРИЧЕСКОГО СОПРОТИВЛЕНИЯ KR=(∆R/∆)100 %, где ∆R − относительное изменение электросопротивления образца. 2.4.2. Влияние внешних факторов на химическую коррозию металлов Скорость и характер процесса химической коррозии металлов зависит от ряда факторов. ВНЕШНИМИ ФАКТОРАМИ называют те, которые связаны с составом коррозионной среды и условиями коррозии (температура, давление, скорость перемещения коррозионной среды и т.д.). Температура − мощный внешний фактор коррозии. Характер влияния температуры на скорость окисления металла определяется зависимостью константы скорости реакции окисления (k) и диффузии (D) от температуры. И k = f(T) и D = f(T) описываются одним и тем же уравнением (уравнение Аррениуса): k = k0 exp (−E/RT), (9) где k0 − константа; Е − энергия активации химической реакции. D = D0 exp (−E1/RT), (10) 1 где D0 – константа; Е − энергия активации диффузии. Таким образом, вне зависимости от вида контролирующей стадии процесса окисления с повышением температуры скорость окисления резко возрастает. Колебания температуры, особенно переменный нагрев и охлаждение, увеличивают скорость окисления металла, так как в защитной пленке образуются трещины. Состав газовой фазы. Влияние состава газовой фазы на скорость коррозии металла велико, специфично и изменяется с изменением температуры. В частности, на скорость окисления железа и 21 стали особенно сильно влияют кислород, соединения серы, водяные пары. Приведенные ниже данные свидетельствуют о зависимости увеличения относительной скорости коррозии стали с 0,17 % углерода от состава газовой фазы при 900 оС: Состав газовой фазы Кислород Чистый воздух Чистый воздух + 2 % SO2 Чистый воздух + 5 % H2O Чистый воздух + 5 % SO2+ 5 % H2O % 200 100 118 134 276 Значительное влияние на коррозию сталей и сплавов оказывают продукты горения топлива, содержащие ванадий (например, V2O5). Это соединение находится в золе от сжигания дешевого топлива − мазута, нефтепродуктов. Зола, налипая на металл, увеличивает скорость его коррозии в десятки раз. Причина этому – «ванадиевая коррозия», обусловленная легкоплавкостью V2O5, и его способностью ОФЛЮСОВЫВАТЬ (переводить в жидкое состояние) химические соединения золы и окалины. Пятиокись ванадия участвует в процессе окисления железа: 4Fe + 3V2O5 = 2Fe2O3 + 3V2O3, V2O3 + O2 = V2O5, V2O5 + Fe2O3 = 2FeVO4. Повышение содержания СО в газовой фазе понижет скорость коррозии углеродистых и низколегированных сталей, но при больших количествах СО в газовой фазе может произойти науглероживание поверхности стали. При этом возможны следующие химические реакции: 2CO + O2 = 2CO2, 2CO = Cсаж + CO2. Давление окислителя. Если бы поверхность металла не была покрыта оксидной пленкой, то скорость окисления была бы пропорциональна парциальному давлению окислителя (для двухатомных газов – PГ2). Так как поверхность металла покрыта слоем оксида, то зависимость скорости окисления от величины парциального давления окисляющего газа определяется строением защитной пленки. В то же время при окислении железа при 700 − 950о С скорость окисления не зависит от парциального давления Po2. 22 Скорость движения газовой фазы. Окисление как гетерогенный процесс определяется скоростью подвода и отвода реагентов в зону реакции. Поэтому, чем больше скорость движения потока газа, тем больше и скорость окисления металла. Режим нагрева металла. Влияние режима нагрева металла может быть рассмотрено в зависимости от влияния колебаний температуры. Иначе говоря, переменные нагрев и охлаждение увеличивают скорость окисления ввиду нарушения сохранности защитной пленки. 2.4.3. Влияние внутренних факторов на химическую коррозию металлов ВНУТРЕННИМИ называют факторы, связанные с составом, структурой, внутренними напряжениями в металле, качеством обработки поверхности металла и др. Состав сплава. Применительно к наиболее важным конструкционным сплавам на основе железа для случая газовой коррозии можно отметить следующее. 1. При высоких температурах (более 800оС) с увеличением содержания углерода в стали скорость ее окисления и обезуглероживания уменьшается вследствие более активного образования СО, что снижает окислительный потенциал газовой фазы. 2. Сера фосфор, никель, марганец практически не влияют на скорость окисления железа. 3.Титан, медь, кобальт, бериллий заметно снижают скорость окисления железа. 4. Хром, алюминий, кремний сильно замедляют окисление железа. 5. Ванадий, вольфрам и молибден могут значительно ускорить окисление стали, которое иногда носит катастрофический характер. Структура сплава. Анализ экспериментальных данных свидетельствует о том, что чем меньше в сплаве структурных составляющих, тем выше его жаростойкость. Применительно к сплавам железо – углерод наиболее устойчивой является АУСТЕНИТНАЯ СТРУКТУРА9. Аустенит (γ-фаза) − высокотемпературная гранецентрированная модификация железа и его сплавов. В углеродистых сталях аустенит − это твѐрдый раствор внедрения, в котором атомы углерода входят внутрь элементарной ячейки γ-железа во время конечной термообработки. В сталях, содержащих другие металлы (кроме железа, легированные стали), атомы металлов замещают атомы железа в кристаллической решетке, и возникает твердый раствор замещения. Фаза названа в честь сэра Уильяма Чандлера Робертс-Остина (англ. William Chandler Roberts-Austen, 1843 − 1902). 9 23 Всего различают пять больших групп нержавеющих сталей, определяемых их микроструктурой. Наиболее распространенными являются три: АУСТЕТНИТНЫЕ (AUSTENITIC) − не магнитная сталь с основными составляющими 15−20 % хрома и 5−15 % никеля, который увеличивает сопротивление коррозии. Она хорошо подвергается тепловой обработке и сварке. Обозначается буквой A в начале наименования марки. Именно аустенитная группа сталей наиболее широко используется в промышленности и в производстве элементов крепежа. МАРТЕНСИТНЫЕ (MARTENSITIC) − значительно более твердые, чем аустетнитные стали и могут быть магнитными. Они упрочняются, закалкой и отпуском подобно простым углеродистым сталям и находят применение главным образом в изготовлении столовых приборов, режущих инструментов и общем машиностроении. Больше подвержены коррозии. Обозначаются буквой С. ФЕРРИТНЫЕ (FERRITIC) стали значительно более мягкие чем мартенситные по причине малого содержания углерода. Они также обладают магнитными свойствами. Обозначаются буквой F. Стали с двухфазной АУСТЕНИТНО−ФЕРРИТНОЙ СТРУКТУРОЙ менее устойчивы против окисления. Их меньшая жаростойкость связывается с большей неоднородностью образующейся защитной пленки, что приводит к ее разрушению при росте (неоднородность возникающих внутренних напряжений). Так хромоникелевые стали с однофазной аустенитной структурой более устойчивы против окисления, чем двухфазные: Х12Н12М2Т, Х12Н9Т ведут себя лучше, чем ОХ21Н5МД2Т или 1Х21Н5Т 2]. На жаростойкость10 чугунов кроме их состава влияет и структура. Существенное влияние оказывает форма графитных включений. Так, при шаровидной форме графита стойкость против окисления выше, чем при пластинчатой. Установлено, что предварительная холодная пластичная деформация несколько ускоряет окисление металла вследствие повышения запаса его энергии. Чем тщательнее обработана поверхность металла, тем меньше скорость его окислеЖаростойкость − окалиностойкость, способность металлических материалов противостоять химическому разрушению поверхности под воздействием воздушной или иных газообразных сред при высоких температурах. 10 24 ния, что обусловлено лучшей сохранностью защитных пленок на гладкой поверхности. 2.5. Некоторые случаи газовой коррозии металлов в технологических средах В химической промышленности многие технологические процессы или их определенные стадии протекают в газовой среде в условиях повышенных температур и давлений (рис.4). При температурах от 100 до 200 − 300 °С многие газы не опасны. Химическая активность газов и скорость газовой коррозии металлов сильно возрастают при температурах выше 200 − 300 °С. Так, Cl2 начинает действовать на железные сплавы при Рис. 4. Некоторые случаи газовой температуре > 200 °C, HCl - > koroze kovů v technologické - 300 °C, SO2, NO2, páry síry. > 500 °C. Tyto vlastnosti chování prostředí technologických plynů a jejich široké využití v průmyslu vyžadují podrobnější zvážení chování kovů v reálných podmínkách. 2.5.1. Koroze železa, litiny a ocelí v atmosféře par O2, CO2, H2O Působením vzdušného kyslíku při vysokých teplotách na slitiny železo-uhlík dochází k oxidaci železa s tvorbou okují, oduhličením oceli a růstem. z litiny. V důsledku oxidace železa za vysokých teplot vzniká vrstva korozních produktů zvaná VODNÍK. Okuje má složitou strukturu a obsahuje několik oxidů: Fe3O4 – MAGNETIT má složitou spinelovou krystalovou mřížku; Fe2O3 – HEMATIT má romboedrickou mřížku; FeO – VUSTIT má vadnou strukturu krystalové mřížky. Wustite vzniká při teplotách nad 575 oC a při pomalém ochlazování se rozkládá: 4FeO Fe3O4 + Fe. 25 Pod 575 °C není v okuje žádný wustit a vrstva Fe3O4 přímo přiléhá k povrchu oceli. Rychlost oxidace oceli (viz oddíl 2.4.1) roste s rostoucí teplotou podle zákona blízkého exponenciále (obr. 5) a v souřadnicích logk − 1/T je vyjádřeno přerušovanou čarou, jejíž každý zlom odpovídá nějakou transformaci. Při teplotě 575 ºC se tak v okuje objeví podvrstva wustitu FeO, která neinterferuje s difúzí kyslíku, v důsledku čehož se zvyšuje aktivační energie procesu a zvyšuje se rychlost oxidace kovu. Přerušení křivky při teplotě ~ 900 ºС odpovídá alotropické přeměně oceli. Charakter změny teplotní závislosti rychlosti oxidace oceli naznačuje, že austenitická struktura je odolnější vůči teplu, u které je pozorován pomalejší nárůst rychlosti oxidace s rostoucí teplotou. Spolu s oxidací v ocelích a litině dochází k procesu dekarbonizace - ochuzení povrchové vrstvy o uhlík v důsledku interakce v nich obsaženého karbidu železa s kyslíkem a činidly obsahujícími kyslík Fe3C+O2→3Fe+CO2 Fe3C+CO2 →3Fe+2CO Fe3C+H2O→3Fe+CO + H2. 26 Složení plynného prostředí má silný vliv na rychlost oxidace oceli a litiny. Při plynové korozi mohou být agresivními činiteli např. chlór, sloučeniny síry, kyslík, vzduch, sloučeniny jódu atd. Jejich agresivita vůči různým kovům není stejná, proto se rychlost koroze liší. Rychlost oxidace Fe, Co, Ni při teplotě 900 °C tedy roste v pořadí H2O → CO2 → O2 → SO2. V tomto případě jsou kovy v závislosti na rychlosti koroze v atmosféře těchto činidel uspořádány ve vzestupné řadě: Ni → Co → Fe. Pokud předpokládáme, že rychlost koroze na vzduchu při 900 °C je 100 %, pak přidání 2 % SO2 tuto rychlost zvýší na 118 %, 5 % H2O na 134 %, 2 % SO2 + 5 % H2O na 276 %. 2.5.2. Koroze pod vlivem produktů spalování paliva Produkty spalování paliva (uhlí, topný olej atd.) ve většině případů obsahují značné množství sloučenin síry (SO2, H2S) a vanadu ve formě V2O5, což je silné oxidační činidlo. Nízkotavitelný oxid vanadičitý interaguje s ochrannou vrstvou vodního kamene na kovovém povrchu, rozrušuje ji a tvoří vanadičnany, které vytvářejí eutektika11 s nízkým bodem tání Fe2O3 + V2O5 → 2FeVO4 s oxidem vanadičným V2O5. Vanadičnan železitý (FeVO4) má tedy bod tání = 840 °C a bod tání jeho eutektika s V2O5 je 625 °C; Teplota tání vanadičnanu chromitého CrVO4 je 810 °C a jeho eutektikum s V2O5 je 665 °C. Poté se vanadičnan aktivně účastní procesu oxidace samotného kovu 6FeVO4+4Fe→ 5Fe2O3 + 3V2O3 4Fe+ 3V2O5→ 2Fe2O3 + 3V2O3 V2O3+O2→ V2O5 a samotný V2O5 se prakticky nespotřebovává. V největší míře koroze vanadu postihuje pecní hady, podpěry a přepážky pracující v teplotním rozmezí 620 – 700 °C. Eutektikum (z řeckého éutektos - snadno se taví), kapalná soustava (roztok nebo tavenina), která je při daném tlaku v rovnováze s pevnými fázemi, jejichž počet se rovná počtu složek soustavy. 11 27 Rychlost koroze i u chromových a chromniklových ocelí, které nejsou náchylné k vysokoteplotní sirovodíkové korozi, přitom dosahuje 15 mm/rok. Vlivem sloučenin síry podléhají železo-uhlíkové oceli intenzivní mezikrystalové korozi v důsledku většího počtu defektů v krystalových mřížkách sulfidů než oxidů. To vede k zintenzivnění difúzních procesů. S nárůstem obsahu oxidu uhelnatého (II) ve zplodinách spalování znatelně klesá rychlost plynové koroze uhlíkových a nízkolegovaných ocelí. Jeho velmi vysoká koncentrace však vede k nauhličování povrchu: 3Fe+2CO→Fe3С+CO2. Vysoce legované slitiny typu X40N50 jsou odolné vůči korozi vanadu. Zavádění křemíku do oceli má také pozitivní vliv na odolnost oceli legování molybdenem, wolframem a vanadem, protože tyto legující prvky podporují12 tvorbu V2O5. Slitiny na bázi Cu jsou k takové korozi vysoce náchylné. Ke snížení rychlosti koroze vanadu se v současnosti používá: − omezení provozní teploty na 650 °C; − použití slitin typu X40N50 pro výrobu součástí podléhajících tomuto druhu destrukce; − foukání do paliva dolomitového prachu obsahujícího oxidy hořčíku a hliníku, které tvoří žáruvzdorné sloučeniny s oxidem vanadu; − omezení celkového obsahu sodíku a vanadu v palivu na maximálně 2 10–4 %. 2.5.3. Koroze v prostředí chlóru a chlorovodíku Chování kovů v prostředí plynného chloru a chlorovodíku se zásadně liší od působení jiných agresivních prostředí. Je to způsobeno tím, že chloridové soli, které se tvoří na povrchu kovu, mají nízkou teplotu tání a v některých případech při zvýšení teploty sublimují13, například Ti + 2Cl2 → TiCl4. Propagace - (synonyma) řízení, propagace, režie. Sublimace je přechod látky z pevného do plynného skupenství, přičemž se obchází kapalná fáze; stejné jako sublimace. 12 13 28 Většina těchto reakcí je exotermická. Rychlost odvodu tepla se ukazuje být nižší než rychlost samotné reakce, v důsledku čehož se kovy vznítí a „hoří“ v atmosféře chlóru. To vede k výraznému lokálnímu zvýšení teploty a vzniklé chloridy tají a rozkládají se. Nejodolnější vůči chlóru jsou niklové, olověné a chromové oceli. Teplota vznícení některých kovů v atmosféře suchého chlóru: o t vznícení. S Ti< 20 Pb 90−100 Fe 150 Cu 200 Ni > 500. V suchém chlorovodíku při pokojové teplotě má řada kovů a slitin uspokojivou odolnost. S rostoucí teplotou odpor kovových materiálů postupně klesá až na teplotu specifickou pro každý kov. Maximální vysoké teploty přípustné při dlouhodobém provozu kovů a slitin v suchém chloru a chlorovodíku jsou uvedeny v tabulce. 2. Nejodolnější kovy v suchém chlóru, s výjimkou ušlechtilých kovů, jsou nikl a jeho slitiny. Povrchové filmy vytvořené na niklových a chromniklových ocelích mají nízkou těkavost a vyhovující ochranné vlastnosti. Tabulka 2 Přípustné teploty při provozu některých kovů a slitin v atmosféře chlorovodíku a chlóru3] T, °C Materiál Cl2 1200 − − 550 − 100 150 300 − Platina Zlato Wolfram Nikl Inconel (80 % Ni, 14 % Cr, 6% Fe) Měď Uhlíková ocel St.Z Nerezová ocel 12Х18Н9Т Stříbro 29 HCl 1200 870 600 510−600 480 100−120 260−350 450−500 230 2.5.4. Vodíková koroze oceli Vodíková koroze může doprovázet mnoho technologických procesů probíhajících při zvýšených teplotách od 200°C a tlacích od 300 MPa v prostředích obsahujících vodík. Tyto podmínky odpovídají procesům, jako je hydrogenace uhlí a ropy, syntéza čpavku a metanolu atd. Jsou pozorovány dva typy poškození kovů vodíkem - VODÍKOVÁ KŘEHKOST a VODÍKOVÁ KOROZE. Často se tyto jevy navzájem překrývají. Pokud je v plynu přítomen čpavek, může dojít i k NITRIDACI KOVU. Když se směs dusíku a vodíku dostane do kontaktu s kovem za podmínek zvýšené teploty a tlaku, molekulární vodík disociuje na povrchu kovu. Vzniklý atomární vodík difunduje do kovové mřížky a rozpouští se v ní. Jak teplota klesá v důsledku snížené rozpustnosti, vodík má tendenci přejít uvnitř kovu do plynného stavu. V tomto případě vznikají v kovu velká napětí, která vedou k nevratné křehkosti. Vodíková koroze je výsledkem chemické interakce vodíku s karbidovou složkou oceli. Navenek znamená projev vodíkové koroze silný pokles pevnosti oceli bez znatelné destrukce povrchu. Mechanismus vodíkové koroze zahrnuje následující fáze: - při vysokých teplotách molekulární vodík disociuje na atomy, které jsou sorbovány povrchem oceli a difundují do její krystalové mřížky; − atomární vodík jako aktivní redukční činidlo redukuje karbid železa a interaguje s uhlíkem rozpuštěným v oceli prostřednictvím nevratných reakcí: Fe3C + 2H2 3Fe + CH4 C + 4H CH4, čímž ocel zbavuje její zpevňující báze. Vzhledem k tomu, že difúzní procesy, včetně pohybu vodíku, se nejsnáze realizují podél hranic zrn, kde jsou převážně umístěny cementitové desky, jejich destrukce vodíkem vede k narušení spojení mezi krystality a tím ke snížení tažnosti oceli. . 30 Výsledný metan má ve srovnání s parametry feritové krystalové mřížky velkou molekulovou velikost. V důsledku toho nemůže difundovat z větší části kovu a hromadí se v jeho mikrodutinách a defektech, což způsobuje vysoký tlak v dutině a vede k praskání. V tomto případě vznikají trhliny podél hranic zrn. U měkkých ocelí s nízkými pevnostními vlastnostmi se akumulace metanu vyskytují převážně v připovrchové vrstvě, vytvářející bobtnání, která jsou jasně viditelná na povrchu oceli. Kromě toho, že se vodík podílí na tvorbě metanu, má dobrou rozpustnost v kovu. Koncentrace vodíku rozpuštěného v kovu závisí na parciálním tlaku vodíku na rozhraní kov-plyn a lze ji určit podle vzorce: = Ks p1/2, kde je množství rozpuštěného vodíku v oceli, cm3/ 100 g; p – parciální tlak vodíku, at; Ks – rozpustnost vodíku v oceli při p = 1 at. Protože rozpouštění vodíku kovem je endotermický proces, rozpustnost vodíku se zvyšuje s rostoucí teplotou. Tento jev se nazývá PROPUSTNOST VODÍKU (VH) 4]. Propustnost vodíku oceli závisí na obsahu uhlíku a legujících prvků v ní a klesá s rostoucím obsahem uhlíku, jak dokládají údaje uvedené v tabulce. 3. Tabulka 3 Vliv obsahu uhlíku na propustnost vodíku uhlíkových ocelí Kov C, hm.% VН, cm3/cm2h mm–1 Technické železo 0,04 5,10 Ocel 15 0,15 4,09 Ocel 30 0,27 2,79 Ocel U10A 1,10 2,24 Neobjevuje se vodíková koroze okamžitě od okamžiku, kdy je ocel vystavena vodíku, ale po určité době. Tato doba, během které nedochází k žádným změnám v mikrostruktuře a mechanických vlastnostech oceli, se nazývá INKUBAČNÍ DOBA PROCESU DEKARBACE OCELI. 31 Inkubační doba má velký praktický význam, protože v podstatě určuje bezpečnou provozní dobu zařízení. Inkubační doba závisí především na teplotě a tlaku. S rostoucí teplotou a tlakem se inkubační doba zkracuje. Délka inkubační doby je přitom značně ovlivněna chemickým složením oceli. Teplota a parciální tlak vodíku ovlivňují nejen dobu trvání inkubační doby, ale také rychlost oduhličení oceli při vodíkové korozi. 2.5.5. Karbonylová koroze Karbonylová koroze se vyskytuje v technologických procesech s uhlíkem za zvýšených tlaků a teplot. Mezi takové procesy patří například výroba methylalkoholu a butylalkoholu, konverze metanu a oxidu uhelnatého. Za normálních podmínek je CO vůči kovům inertní. Při vysokých teplotách a tlacích oxid uhelnatý reaguje s mnoha kovy a tvoří karbonyly. Například Fe + nCO = Fe(CO)n. Železo a CO mohou tvořit tři sloučeniny: tetrakarbonyl - Fe(CO)4, pentakarbonyl - Fe(CO)5 a nanokarbonyl - Fe(CO)g. Všechny tyto sloučeniny jsou značně nestabilní a rozkládají se, když teplota stoupá. Nejstabilnější sloučeninou z nich je Fe(CO)5. Tvorba karbonylů se zvyšuje se zvyšující se teplotou od 100 ºС, poté je v teplotním rozmezí 140 – 180 ºС pozorována jejich téměř úplná disociace na Fe a CO. Oxid uhelnatý (II) může tvořit podobné sloučeniny s mnoha dalšími kovy. Karbonylová koroze způsobuje destrukci a uvolnění povrchové vrstvy kovu do hloubky 5 mm. Ke změnám struktury kovu ve větší vzdálenosti od povrchu již nedochází. 2.5.6. Koroze v neelektrolytech NEELEKTROLYTY jsou kapaliny, které nevedou elektrický proud. Kapalná korozivní média anorganického původu - kapalný brom, roztavená síra apod. Kapalné organické látky - benzen, chloroform aj., kapalná paliva (olej, petrolej, benzín aj.), mazací oleje. V čisté formě jsou málo agresivní, nicméně přítomnost i malého množství nečistot v nich (merkaptany14, sirovodík, voda, kyslík atd.) prudce zvyšuje jejich chemickou aktivitu. Merkaptany a sirovodík obsažené v ropě tak způsobují korozi Fe, Cu, Ni, Ag, Pb, Sn za vzniku jejich merkaptidů (Me−(S−R)n) a polysulfidů (Me2Sx). Stopy vody v tetrachlormethanu zvyšují jeho korozivitu v důsledku tvorby vodivých složek (HCl) v důsledku hydrolýzy a vzniku koroze elektrochemickým mechanismem: CCl4 + H2O → COCl2 + 2HCl CCl4 + 2H2O + kаt → CO2 + 4HCl Především koroze kovů v neelektrolytech probíhá chemickým mechanismem. Tento proces se skládá z několika stupňů, z nichž každý určuje rychlost koroze: 1) difúze oxidačního činidla na kovový povrch; 2) chemisorpce oxidačního činidla; 3) chemická reakce; 4) desorpce korozních produktů z kovového povrchu; 5) difúze korozních produktů do objemu neelektrolytu. V některých případech se na kovovém povrchu vytvoří ochranný film z korozních produktů, což vede k inhibici procesu koroze v důsledku obtížnosti difúze okysličovadla na kovový povrch. V závislosti na ochranných vlastnostech výsledného filmu a jeho rozpustnosti lze instalovat difúzní, kinetické nebo smíšené difuzně-kinetické řízení procesu. Teplota významně ovlivňuje rychlost chemické koroze kovů v neelektrolytech (viz část 2.4.). Vliv teploty na rychlost procesu je v některých případech komplikován změnami rozpustnosti oxidačního činidla a filmu kovových korozních produktů v neelektrolytu při změně teploty. 14 Merkaptany jsou hydrosulfidové deriváty uhlovodíků: R – S –H. 33 Přítomnost vody v neelektrolytech výrazně aktivuje korozní účinek nečistot a způsobuje intenzivní elektrochemickou korozi kovů, tzn. mění se mechanismus korozního procesu. K ochraně kovů v neelektrolytech před chemickou korozí se vybírají kovy a slitiny, které jsou v daném prostředí stabilní (například hliník a jeho slitiny jsou stabilní v krakovaném benzínu), aplikují se ochranné nátěry (například ocel v sirovodíkové prostředí je potaženo hliníkem). 2.6. Způsoby ochrany kovů před různými typy plynové koroze 2.6.1. Metody ochrany oceli před korozí plynem K ochraně oceli před korozí plynem se nejrozšířeněji používají tepelně odolné NÁTĚRY PŘILEPOVACÍ A OCHRANNÉ. Slévání - (německy legieren - „spojovat“, z latinského ligare – „vázat“) – přidávání nečistot do složení materiálů za účelem změny (zlepšení) fyzikálních a/nebo chemických vlastností základního materiálu. Hlavní metody ochrany kovů před oxidací při vysokých teplotách jsou založeny na SLEVÁNI. Odolnost kovů oxidovaných při vysokých teplotách závisí na ochranných vlastnostech oxidového filmu pokrývajícího kov. Prvky, které přispívají k vytvoření ochranné vrstvy na slitinách železa a uhlíku, jsou chrom, hliník a křemík. Tyto prvky oxidují snadněji při vysokých teplotách na vzduchu než legovaný kov a tvoří stabilnější okují. Podle teorie žáruvzdorného legování tvoří legující složka na povrchu kovu ochrannou vrstvu tvořenou pouze oxidem legovací složky. Legující prvek musí splňovat následující základní požadavky: Oxid vytvořený na povrchu kovu musí být spojitý, tzn. poměr Vok/VMe musí být >1. Vrstva oxidu musí mít vysoký ohmický odpor rMeO > rFe, velikost iontů legující složky musí být menší než velikost iontů základního kovu, tzn. rMe< rFe. Это условие вытекает из следующих соображений: 34 а) меньший радиус иона легирующего компонента по сравнению с ионом основного металла позволяет предполагать у легирующего компонента больший коэффициент диффузии в сплаве, что может обеспечить преимущественный выход ионов компонента на поверхность сплава и, следовательно, преимущественное образование оксида легирующего компонента; б) меньший радиус иона легирующего компонента позволит образовать оксид с соответствующими меньшими параметрами, который будет оказывать большое сопротивление диффузии через него и, следовательно, создавать большое затруднение для окисления основного металла. Это подтверждается сравнением размеров ионных радиусов железа и легирующих элементов: хрома, алюминия и кремния Ион металла Ионный радиус, Аº Fe2+ 0,74 Cr6+ 0,52 Al3+ 0,50 Si4+ 0,41. А энергия образования оксида легирующего компонента должна быть больше энергии образования оксида основного металла ΔGMeO<ΔGFeO. Если это условие не соблюдается, то оксид легирующего компонента будет восстанавливаться основным компонентом, как например, оксидная пленка меди, которая не может быть устойчивой на железном сплаве вследствие возможности протекания реакции CuO+Fe → FeO+Cu. При этом необходимо соблюдение условий: высокая температура плавления оксида легирующего компонента, низкая упругость диссоциации, а также отсутствие низкоплавких эвтектик в смеси с другими оксидами для того, чтобы оксид легирующего компонента был достаточно устойчив. В качестве примера можно привести элемент бор, который является аналогом Al по таблице Менделеева, но дает легкоплавкие оксиды. Температура плавления ВО3 − 294 0С, и поэтому бор, в отличие от алюминия, не может быть легирующим компонентом, повышающим жаростойкость. Необходимым условием является также, образование твердых растворов между легирующими компонентами и основным металлом, так как при этом условии возможно равномерное распределение этого элемента на поверхности и образование сплошной пленки оксида легирующего компонента. 35 Существует и другая теория, согласно которой легирующий элемент образует на поверхности сплава смешанные оксиды, обладающие повышенными защитными свойствами, по сравнению с оксидами чистых компонентов. Механизм повышения жаростойкости можно свести к тому, что легирующий компонент должен уменьшить возможность образования в окалине вюститной фазы (FeO) на стали и, таким образом, благоприятствовать образованию шпинельной фазы типа Fe3O4ּγFe2O3 с возможно меньшим параметром решетки. Жаростойкие легированные стали имеют на поверхности оксидные слои со структурой именно шпинели. Еще более высокими защитными свойствами обладают сложные шпинели типа FeOּMe2O3 или Fe2O3 ּMeO. С целью экономии легированных сталей жаростойкость углеродистых сталей повышают, применяя защитные покрытия. Для нанесения на сталь защитных покрытий пользуются следующими методами: ТЕРМОДИФФУЗИОННЫМ, НАПЛАВКОЙ И ПЛАКИРОВАНИЕМ. Для нанесения покрытий термодиффузионным методом стальное изделие после очистки поверхности механическим способом и травлением помещают в реакционную смесь, состоящую из порошка наносимого металла и катализатора. Например, при нанесении алюминиевых покрытий смесь содержит измельченный ферроалюминиевый и хлористый аммоний. Процесс насыщения поверхности стали алюминием (АЛИТИРОВАНИЕ) производится при высоких температурах (900−9500С), в результате чего происходит разложение хлористого аммония на аммиак и хлористый водород: NH4Cl → NH3 + HCl, который, в свою очередь, взаимодействует с алюминием 2Al +6HCl → 3H2 + 2AlCl3. На поверхности стального изделия происходит выделение атомарного алюминия AlCl3 +Fe → FeCl3 + Al, который диффундирует в сталь. Значительное повышение жаростойкости стали термодиффузионным покрытием обусловлено образованием окислов Al2O3 или двойных окислов FeAl2O4 в алитированном слое. На поверхности хромированных или силицированных изделий образуются соответствующие оксиды хрома или кремния. 36 НАПЛАВКА − это нанесение слоя металла или сплава на поверх- ность изделия посредством сварки плавлением. ПЛАКИРОВА́НИЕ − (фр. plaquer − накладывать, покрывать) термомеханическое покрытие − нанесение на поверхность металлических листов, плит, проволоки, труб тонкого слоя другого металла или сплава термомеханическим способом. Для защиты от карбонильной коррозии применяют хромистые стали с содержанием 30 % Сг, хромоникелевые стали с содержанием 23 % Сг и 20 % № и марганцевые бронзы доя работы при температуре до 700 °С и давлении до 35 МПа. При более низких параметрах возможно применение менее легированных сталей типа Х18Н9. Сырьем для синтеза мочевины CO(NH2)2 является NH3 и СО2. Процесс протекает при температуре 175−190°С и давлении 20 МПа. Хромистые нержавеющие стали различных марок непригодны для изготовления основных аппаратов. Наибольшую стойкость имеют стали, легированные молибденом, и хромникельмолибденовомедные стали. Важным фактором для повышения коррозионной устойчивости является тщательная очистка газов от сероводорода и дополнительное введение в систему кислорода в количестве 0,5- 1,0 об.% от содержания СО2. При сернистой коррозии сера и ее соединения - сернистый ангидрид (SO2), сероводород (H2S), меркаптаны или тиоспирты и т.д. являются достаточно агрессивными, коррозионно−активными веществами. Наиболее активным компонентом при высокотемпературной газовой коррозии является сероводород. Он даже более опасен, чем диоксид серы. Сернистый газ SO2 является исходным продуктом при производстве серной кислоты. Его получают при обжиге серного колчедана, сжигании серы, из сероводорода при утилизации отходящих газов металлургических производств. Чугунные детали скребков конверторных печей кипящего слоя, зубья и гребки колчеданных печей, котлы−утилизаторы, сухие электрофильтры, газоходы обжиговых газов в производстве серной кислоты часто выходят из строя вследствие газовой коррозии. В результате коррозии черных металлов в сернистом газе при температурах 300°С и выше образуется слоистая окалина, состоящая из FeS, FeO и др. 37 При температуре газа более 400 °С для деталей из чугуна характерно увеличение объема металла, достигающего 10 % от начальной величины. При этом резко снижается прочность материала. Детали испытывают коробление, трескаются и разрушаются. Это явление называется «ростом» чугуна и объясняется внутренним окислением металла. Максимальный рост чугуна наблюдается при 700 °С. К ростоустойчивым чугунам относятся высоколегированные хромистые чугуны, карбидный чугун типа «пирофераль» и «чугаль» Сернистый газ при высоких температурах окисляет никель. При этом образуется окалина, в состав которой входят NiS и NiO: 3Ni + S02 = NiS + 2NiO. Сернистый никель образуется и при действии на металл сероводорода: Ni + H2S = NiS + Н2. Сульфид никеля с металлическим никелем образует легкоплавкую эвтектику с температурой плавления около 625 °С. Образование этой эвтектики в сталях, содержащих никель, происходит преимущественно по границам зерен, вызывая разрушение металла. Стали с содержанием никеля выше 15 % очень чувствительны к действию сернистого газа. В процессе окисления они теряют механическую прочность. Поэтому при работе с газовой средой, содержащей диоксид серы, при температурах до 400 °С используют углеродистые стали, а при более высоких температурах − хромистые стали. Наиболее употребительны жаростойкие стали − 4Х9СА, Х6СЮ, XI7, ОХ 17 Г, X1800, Х25Т. Интенсивное образование окалины происходит при температурах выше 800−1000 °С. К жаропрочным сталям в этой среде относятся Х5М, Х6СМ, XI8Н12Т, Х23Н18. Рабочая температура для этих сплавов 550−600 °С (для Х23Н18 − 1000 °С). Сухой сернистый газ реагирует с алюминием очень медленно. Поэтому алюминий используют для защиты от коррозии деталей и узлов теплообменников и контактных аппаратов. Сухой сероводород при комнатной температуре не представляет опасности для обычных углеродистых сталей. С повышением температуры опасность сероводородной коррозии углеродистых сталей значительно увеличивается. При температуре выше 300 °С 38 железо подвергается сильной коррозии в серосодержащих газовых средах. Легирование хромом в количестве > 12 % zvyšuje odolnost proti korozi při teplotách do 700–800 °C. Při korozi chromových ocelí se tvoří okuje, jehož vnější vrstva se skládá ze sulfidu železa. V této vrstvě není prakticky žádný chrom. Veškerý oxidovaný chrom je koncentrován ve vnitřní vrstvě, která má ochrannou vlastnost. Feritické Obr. 5 10 15 20 mají dobrou chemickou odolnost v atmosféře síra-Cr, hm. % vodíku. 6. Rychlost koroze chromových slitin obsahujících 25–30 % pevné oceli v olejových parách při 650 °C. Čísla na křivkách označují chrom. Obsah H2S, % Zvláště nebezpečná je kombinovaná přítomnost sloučenin síry a dalších žíravých složek. V ropném průmyslu tak představuje při tepelném zpracování sirných olejů zvláštní nebezpečí směs sirovodíku a vodíku. Z těch zobrazených na Obr. 6 údaje ukazují, že rychlost koroze chromových ocelí se zvyšuje se zvyšující se koncentrací sirovodíku v olejových parách. V tomto případě 10násobné zvýšení koncentrace H2S způsobí zvýšení rychlosti koroze více než 12–15krát. Při hoření paliva vznikají složité směsi plynů obsahující O2 a různé oxidy, včetně sirných nečistot. V těchto případech je pozorována koroze SULFIDE-OXID. Ochranný film na kovu se obvykle skládá z několika vrstev. Vnější vrstva je obohacena kyslíkem a skládá se z oxidu kovu a vnitřní vrstvy přiléhající k povrchu kovu obsahují zvýšené množství síry a sulfidů. Pokud při spalování paliva vzniká popel, který obsahuje oxid vanadičitý V2O5, pak se rychlost koroze velmi rychle zvyšuje. Příčiny vanadové koroze ocelí byly diskutovány dříve (viz část 2.5.2). 39 Chromové oceli s obsahem 4−6 % Cr jsou považovány za položáruvzdorné. Oceli této třídy jsou díky své dostupnosti, zvýšené korozivzdornosti a pevnosti široce používány v ropném průmyslu pro výrobu krakovacích jednotek. Tepelná odolnost těchto ocelí na vzduchu a ve spalinách s významným obsahem sloučenin síry při teplotách 500–600 ºС je přibližně 3x vyšší než tepelná odolnost nelegovaných ocelí. 2.6.2. Způsoby ochrany proti vodíkové korozi Jedním z hlavních způsobů, jak zvýšit odolnost ocelí vůči vodíku, je SLITOVÁNÍ silnými karbidotvornými prvky, které tvoří stabilnější karbidy než cementit. Takovými prvky jsou chrom, wolfram, molybden, vanad, niob, titan. Typ karbidové fáze má velký vliv na odolnost oceli vůči vodíku. U ocelí legovaných chromem klesá odolnost vůči vodíku v závislosti na složení karbidové fáze v následujících řadách: (Cr,Fe)23C6(Cr,Fe)23C6 + (Cr,Fe)7C3(Cr,Fe) 7C3 (Cr,Fe)7C3 + (Fe,Cr)3C(Fe,Cr)3C Výše uvedená řada ukazuje, že největší odolnosti vůči vodíku u chromových ocelí je dosaženo při tvorbě karbidů (Cr,Fe). ) typ 23C6. Ke vzniku tohoto typu karbidu s obsahem uhlíku v oceli 0,05 % dochází, když je v oceli alespoň 6 % chrómu a se zvýšením obsahu uhlíku je nutné zvýšení obsahu chrómu. Při legování oceli silnějšími karbidotvornými prvky než je chrom, se největší odolnosti oceli vůči vodíku dosáhne, když jsou tyto prvky obsaženy v množství dostatečném k navázání veškerého uhlíku na karbidy typu MeC. V případech, kdy prvky zařízení pracují v podmínkách vysokých úrovní mechanického zatížení, musí mít oceli odolné vůči vodíku také tepelnou odolnost. Pro tyto podmínky se používají oceli s 3 % Cr, navíc legované Mo, V nebo W, například ocel třídy 20Х3МВФ (EI579). Oceli jakosti X5M, X5VF, X9M, 1X13 mají ještě větší odolnost. V nejnáročnějších podmínkách se používají austetické žáruvzdorné chromniklové oceli 0Х18Н10Т nebo Х17Н17М2Т. 40 Další široce používanou metodou zvýšení odolnosti ocelí vůči vodíku je PLÁTENÍ kovy, které mají nižší propustnost vodíku než základní kov. To umožňuje snížit koncentraci vodíku na rozhraní mezi základním kovem a krycí vrstvou a snížit jeho agresivní účinek na základní kov. Výpočet efektivního tlaku vodíku na rozhraní základního a povlakového kovu se provádí podle rovnice: 1/2 1/2 p2 = p1 (11) + kde p1 je tlak vodíku na vnějším povrchu obkladová vrstva; p2 je efektivní tlak vodíku na rozhraní povlak-obecný kov; = VН1/VН2 je poměr propustnosti vodíku obkladové vrstvy a základního kovu; = l1/l2 – poměr tloušťky obkladové vrstvy a základního kovu, resp. Aby se zvýšila propustnost vodíku, mohou být kovy a slitiny uspořádány v řadě: hliník, měď, nikl, X18N10T, 0X13. V praxi se v chemickém a ropném průmyslu používají především bimetaly vyrobené z uhlíkových nebo nízkolegovaných ocelí s ochrannou vrstvou ocelí 0H13 nebo Х18H10Т. Provozní teplota zařízení vyrobeného z vybrané oceli by měla být udržována o 25 °C pod teplotou, při které je možná vodíková koroze. Kapitola 3. ELEKTROCHEMICKÁ KOROZE KOVŮ 3.1. Elektrodové potenciály kovů Při ponoření kovu do elektrolytu dochází na fázovém rozhraní k potenciálovému skoku v důsledku vytvoření dvojité elektrické vrstvy 5, 6. Pokud je kovová deska, například měděná deska, ponořena do vody nebo roztoku měděné soli, pak se kladně nabité ionty Cu2+ začnou pohybovat z kovové vrstvy umístěné na hranici s vodou do vody. V tomto případě se v krystalové mřížce kovu objeví přebytek elektronů a deska získává záporný náboj. Mezi záporně nabitou deskou a kladnými ionty, které prošly do roztoku, vzniká elektrostatická přitažlivost, která brání dalšímu přechodu iontů mědi do roztoku, to znamená, že se proces rozpouštění kovu zastaví. Současně se vyvíjí opačný proces: ionty mědi z roztoku se přibližují k povrchu desky, přijímají z ní elektrony a přecházejí do neutrálního stavu. Po určité době se ustaví stav dynamické rovnováhy, ve kterém je rychlost přechodu iontů z kovu do roztoku rovna rychlosti vypouštění iontů z roztoku na kov. Popsaný jev je schematicky znázorněn na Obr. 7 (ionty kovů jsou pro jednoduchost zobrazeny nehydratované). Rovnováha mezi ionty v roztoku a kovem pro uvažovaný příklad je vyjádřena rovnicí: Cu2+р−р + 2ē Cuºkrystal V rovnovážné rovnici elektrochemické reakce se elektrony obvykle zapisují na levou stranu. tj. proces redukce je zapsán. Obr.7. Schéma vzhledu dvojité elektrické vrstvy na rozhraní kov-roztok Jak je vidět z výše uvedeného příkladu, když kov přijde do kontaktu s roztokem své soli, dvě kontaktní fáze získávají opačné náboje. V důsledku toho se na fázovém rozhraní vytvoří dvojitá elektrická vrstva a mezi kovem a roztokem dojde k potenciálnímu skoku (). Ponoří-li se do roztoku dvě kovové desky z různých kovů (M1 a M11), vznikne galvanický pár, ve kterém se méně aktivní kov redukuje a aktivnější oxiduje (obr. 8). Rýže. 8. Schéma činnosti galvanického článku Obecně je práce tohoto galvanického páru určena rozdílem potenciálů (emf galvanického článku) a je doprovázena bilancí polovičních reakcí: E = ox − red, kde ox je potenciál okysličovadla, V; red – potenciál redukčního činidla, V. Men+(ox) + ne ⇆ Меº Meº(červená) − ne ⇄ Меn+. Když se počet kationtů procházejících do roztoku za jednotku času rovná počtu kationtů uložených na povrchu kovu, dojde k dynamické rovnováze a proces rozpouštění se zastaví. Proto je za takových podmínek nemožný přechod velkého počtu atomů kovových iontů do roztoku. Pokud je však rovnováha elektrické dvojvrstvy narušena vybíjením elektronů nebo odstraňováním atomů kovových iontů, proces koroze bude probíhat bez zábran. Elektrodové potenciály kovů, které jsou v rovnováze s vlastními ionty, se nazývají rovnovážné. Hodnotu rovnovážného potenciálu lze vypočítat pro jakoukoli iontovou aktivitu pomocí Nernstovy rovnice: E = Eº + (RT /nF)ln(aMen+), (12) n+ kde Eº je standardní rozdíl potenciálu při aMe = 1; R – univerzální plynová konstanta, 8,3 kJ/kmolK; T – absolutní teplota, ºK; n – valence kovu; F – Faradayovo číslo 96500 C/mol; 43 aМen+ – aktivita kovových iontů v mol/l. Pokud dosadíme všechny konstanty při 25°C (T = 298 K) a vynásobíme 2,3, abychom se dostali od přirozených logaritmů k desítkovým, dostaneme následující výraz E = Eº + (0,0592/n)log(aMen+), ( 13) Když se roztok zředí, potenciál kovu se posune na zápornou stranu. Je-li např. aktivita iontů Zn2+ v roztoku zinečnaté soli 10–2 mol/l, pak bude rovnovážný potenciál zinku ponořeného v tomto roztoku roven: E = − 0,76 + (0,0592/2) lg10−2 = − 0,819 V, (14) S nárůstem koncentrace kovových iontů v roztoku se potenciál kovu naopak posouvá kladným směrem. Vezmeme-li tedy aktivitu iontů zinku 10 mol/l, pak potenciál zinku bude E = − 0,76 + (0,0592/2)log10 = − 0,73 V, (15) Elektrodové potenciály kovů, ve kterých výměnný proces, který určuje potenciál, zahrnuje nejen jeho vlastní, ale i další ionty a atomy, které se nazývají nerovnovážné nebo nevratné. Pro nerovnovážné potenciály není Nernstův vzorec použitelný, protože reakce probíhající na kovu, tzn. ke ztrátě a akvizici elektronů dochází různými způsoby a potenciál nemůže charakterizovat počátek rovnováhy jedné reakce na elektrodě. Velikost nerovnovážných potenciálů je ovlivněna povahou elektrolytu, teplotou, pohybem elektrolytu, koncentrací roztoku atd. Například potenciál hliníku ve 3% NaCl je − 0,6 V, v 0,05 M Na2SO4 − 0,47 V, a v 0,05 M Na2SO4 + H2S − 0,23 V. V důsledku elektrochemické heterogenity povrchu kovu nebo elektrolytu se na kovu tvoří oblasti s různými potenciály. Hlavní příčiny elektrochemické heterogenity kovového povrchu mohou být následující: − LIKVACE − (La liquation, Saigerung) - je schopnost slitin rozpadat se při přechodu z kapalného do pevného stavu na složky nebo jednotlivé sloučeniny, které mají různé teploty tání. ; − heterogenita ochranného filmu; 44 – heterogenita fyzikálních podmínek – nestejná teplota v různých oblastech kovu, přítomnost deformovaných oblastí, koncentrátory napětí; − heterogenita elektrolytu - rozdíl v koncentraci kyslíku nebo koncentrace soli, rozdílná hodnota pH. Oblasti s negativnějším potenciálem se nazývají ANODICKÉ, oblasti s pozitivnějším potenciálem se nazývají KATODA. Stupeň heterogenity kovového povrchu je určen rozdílem potenciálů katodové a anodové sekce. 3.2. Termodynamika elektrochemické koroze Možnost samovolného vzniku korozního procesu je dána poklesem volné energie (izobaricko-izotermický potenciál) G< 0: G = −nFE < 0, (16) где n – эквивалентное число электронов; Е = К – А – эдс гальванического элемента, образующегося при контакте металла с окислителем в процессе коррозии; К – обратимый потенциал катодной реакции (окислителя); А – обратимый потенциал анодной реакции (металла – восстановителя); F – число Фарадея. Принципиальная возможность протекания процесса электрохимической коррозии металла имеет место при К > A nebo E > 0. Reverzibilní redox potenciál oxidačního činidla v elektrolytu musí být za těchto podmínek kladnější než reverzibilní potenciál kovu za stejných podmínek. Pro stanovení limitů termodynamické možnosti elektrochemické koroze kovů lze použít Pourbaixovy diagramy. Diagramy jsou grafickým znázorněním závislosti reverzibilních elektrodových potenciálů (ve voltech na vodíkové stupnici) na pH roztoku pro rovnovážné stavy: za účasti elektronů - vodorovné čáry; za účasti elektronů a iontů H+ nebo OH– - šikmé čáry; za účasti iontů H+ a OH–, ale bez účasti elektronů (hodnota pH tvorby hydrátu) – svislé čáry. 45 Obr.9. Diagram E – pH pro systém Al-H2O při 25°C Jako příklad na Obr. Obrázek 9 ukazuje Pourbaixův diagram pro systém Al – H2O. Každá oblast diagramu odpovídá jednomu termodynamicky stabilnímu stavu: kovovému stavu ve formě iontů v roztoku, ve formě oxidů nebo hydroxidů. Protože ustavení rovnováhy v roztoku nezávisí pouze na vodíkových iontech, ale i na jiných iontech, je vymezení oblastí tvořeno několika rovnovážnými čarami. Každá z těchto čar odpovídá specifické aktivitě odpovídajících iontů. V oblasti ve spodní části diagramu je hliníkový kov termodynamicky stabilní a nekoroduje. V levé části nad vodorovnou čarou je stav hliníku ve formě Al3+ iontů v roztoku termodynamicky stabilní. Střední oblast odpovídá stabilitě pevných fází oxidu Al2O3 nebo hydroxidu Al(OH)3, které během procesu koroze vytvářejí na povrchu hliníku ochranný film. Pravá strana charakterizuje podmínky, za kterých bude hliník podléhat korozi za vzniku aniontu AlO2– v roztoku. 3.3. Mechanismus elektrochemické koroze Ke korozi kovů v elektrolytech dochází za vzniku galvanických párů, které se nazývají korozní páry. Pokud spustíte zinkové a železné pláty do nádoby s roztoky chloridů zinku a železa (podobně jako diagram pH me na obr. 8) a propojíte je vnějším vodičem, pak miliampérmetr připojený k obvodu ukáže přítomnost elektrického proudu v obvodu. Vznikne galvanický pár, ve kterém zinek oxiduje a přechází do roztoku (anoda). Železo nebude oxidovat a přecházet do roztoku, protože v kombinaci se zinkem působí jako katoda. Železo, nekombinované se zinkem, v podobném roztoku koroduje. V praxi slitiny obsahují velké množství mikroprvků a různých kovů. Jinými slovy, rozdílné kovy jsou umístěny ve stejné rovině v jejich přímém kontaktu mezi sebou navzájem as elektrolytem. Výsledkem je multielektrodový článek, ve kterém se anody střídají s katodami. Na anodických místech přecházejí kovové ionty (Men+) do roztoku. Uvolněné elektrony (nē) se pohybují kovem od anody ke katodě. Rozpouštění kovových iontů ve vodných roztocích by mělo být reprezentováno jako výsledek interakce iontů - atomů umístěných na vnějším povrchu kovu s polárními molekulami vody. Interakce rozpuštěných kovových iontů s vodními dipóly je způsobena silami elektrostatické přitažlivosti. V důsledku toho se kolem každého iontu ve větší či menší míře (v závislosti na velikosti jeho náboje) vytvoří slupka vodních dipólů (fenomén hydratace). Vlivem hydratace se zvětšuje efektivní poloměr iontu, v důsledku čehož výrazně klesá pohyblivost hydratovaných iontů. Proces hydratace je doprovázen uvolňováním energie, zatímco proces dehydratace energii vyžaduje. Kromě vodních dipólů může být iont pokryt obalem jiných dipólů. Obecněji se tento jev nazývá solvace15. Elektrochemická koroze se tedy skládá z následujících fází, probíhajících paralelně: - anodický proces, který spočívá v přechodu kovových iontů z povrchu do roztoku a jejich hydrataci: SOLVACE - proces tvorby asociátů mezi rozpouštědlem a rozpuštěným látky, produktem solvatace je SOLVATE. Pokud je rozpouštědlem voda, pak HYDRATACE, produktem hydratace je HYDRÁT. 15 47 Meº − nē Muži+ + mH2O Muži+ mH2O. hydrát je katodický proces, který spočívá v asimilaci (zachycení) elektronů některým depolarizátorem (D): D + nē . Protože anodické a katodické procesy jsou nezávislé a probíhají snadněji: anodické v oblastech s negativnějším počátečním potenciálem povrchu a katodické v pozitivnějších - a v praktických podmínkách je k tomu nezbytná elektrochemická heterogenita kovového povrchu tyto procesy probíhají převážně lokalizovaným způsobem. Tok elektrického proudu se uskutečňuje na kovu - pohybem elektronů z anodických oblastí ke katodě a v roztoku současně dochází k pohybu iontů. Síla proudu tedy může sloužit a slouží jako kritérium pro rychlost elektrochemické koroze. Vypočítejte spotřebu materiálu anody, tzn. množství zkorodovaného kovu z 1 cm2 jeho povrchu lze použít pomocí Faradayova vzorce: K = Q A / F n = i A / F n, kde K je množství zkorodovaného kov, g/cm2; Q je množství elektřiny protékající během času τ, [s] mezi anodovou a katodovou částí; i – proudová hustota, A/cm2; F – Faradayovo číslo; n – valence kovu; A je atomová hmotnost kovu. 3.4. Polarizace elektrodových procesů a její příčiny Při zkratu elektrod korozního prvku dochází k výrazné změně elektrodových potenciálů, ale odpor dvojice se prakticky nemění. V tomto případě je pozorován pokles elektromotorické síly korozního prvku. Změna počátečních potenciálů vedoucí ke snížení korozního proudu a následně rychlosti koroze se nazývá POLARIZACE. Změna potenciálů (16) elektrod během činnosti korozního prvku ukazuje, že katodový potenciál se stává zápornějším (POLARIZACE KATODY) a potenciál anody se stává kladnějším (ANODICKÁ POLARIZACE): K = Ko – K, (17) o A = A + A, (18) kde K a A jsou hodnoty potenciálu elektrod pracovního prvku; Кº a Аo – počáteční hodnoty potenciálů elektrod před uzavřením obvodu; K a A – potenciální posunutí (polarizace) katody a anody. Rozdíl potenciálů E = K − A se tedy zmenšuje. POLARIZACE je důsledkem zpoždění elektrodových procesů od toku elektronů ve zkratovaném prvku. Ohmické odpory (R) mají malý vliv na snížení korozního proudu, protože jsou obvykle malé. Velký význam mají polarizační odpory, které jsou spojeny buď s obtížemi při výboji elektronů na katodě (Pk), nebo s obtížemi při přechodu kladných iontů kovu z kovové mřížky do roztoku (PA). Tyto odpory, které mají ohmický rozměr, jsou hlavním důvodem poklesu rychlosti elektrochemické koroze. Síla proudu v okamžiku uzavření obvodu podle Ohmova zákona je určena vzorcem Iinit = (Кº − Аo)/R, (19). Počáteční hodnota korozního proudu (Iinit) je přímo úměrná rozdílu mezi počátečními hodnotami potenciálu Kº a Ao za předpokladu, že Kº > Ao, a nepřímo úměrná odporu. Korozní proud v ustáleném stavu (Ipracovní článek) je určen rovnicí: Ipracovní článek = (K – A)/(R + PA + PK). Takže pracuji. el-ta< Iнач. Снижению скорости электрохимической коррозии или уменьшению коррозионного тока могут способствовать: − уменьшение степени термодинамической стабильности, т. е. сближение равновесных потенциалов анодного Аº и катодного Кº процессов; − торможение катодных процессов, иначе говоря, увеличение катодной поляризуемости РК; 49 − торможение анодных процессов, то есть увеличение анодной поляризуемости РА. Поляризация является весьма желательным явлением в коррозионных процессах, так как электроды замкнуты накоротко, омическое сопротивление их невелико и, следовательно, не будь поляризационных сопротивлений, коррозия протекла бы весьма интенсивно. Всякое воздействие, уменьшающее поляризацию, увеличивает скорость коррозионных процессов. Факторы, уменьшающие поляризацию электродов коррозионного элемента, называют ДЕПОЛЯРИЗАТОРАМИ, а процессы, протекающие при этом – АНОДНОЙ И КАТОДНОЙ ДЕПОЛЯРИЗАЦИЕЙ. 3.4.1. Анодная поляризация Анодная поляризация обычно оказывает меньшее влияние на изменение разности потенциалов элемента по сравнению с катодной поляризацией. Анодный процесс, заключающийся в ионизации металла и гидратировании ионов по уравнению: Men+ + mH2O → Men+ ∙ mH2O, может протекать беспрепятственно только при условии беспрерывного отвода образующихся ионов из прианодной зоны. Торможение анодного процесса будет иметь место и в случае отставания скорости выхода ионов металла в раствор от скорости перетекания электронов на катодные участки. В результате анодной поляризации отрицательный заряд на металлической обкладке двойного слоя уменьшается, а следовательно, потенциал металла сдвинется в положительную сторону (или станет менее отрицательны). Таким образом, анодная поляризация происходит или в связи с тем, что скорость анодного процесса не поспевает за скоростью отвода электронов (перенапряжение ионизации металла), или потому, что из−за недостаточно быстрого отвода перешедших в раствор ионов металла повышается концентрация этих ионов в прианодной зоне (концентрационная поляризация). Чем легче протекает анодный процесс, тем меньше его поляризуемость. Следить за тем, легко ли протекает анодный процесс или он заметно тормозится, можно по ходу поляризационной анодной кри50 Потенциал, В вой. Эта кривая показывает зависимость потенциала электрода от плотности анодного тока на нем. Такая кривая показана на рис. 10. Удаление продуктов анодной реакции вызывает деполяризацию анода. Деполяризация анода может быть ускорена перемешиЕ Е ванием раствора, образованием о А таких продуктов коррозии, которые выпадают в осадок, а также путем образования комплексных соединений. Вследствие связывания в комплекс ионов металла, перешедших в раствор, концентрация их в прианодной зоне уменьшаПлотность тока, мА/см2 ется; анодная поляризация сниРис. 10. Анодная поляризаци- жается и переход ионов в расонная кривая твор, т.е. коррозия, усиливается. Например, медь и ее сплавы образуют в растворе аммиака хорошо растворимый комплексный ион 2+. Этим объясняется сильная коррозия меди и ее сплавов в аммиачных растворах. Поляризация анодного процесса в обычных условиях происходит в незначительной степени. Исключение представляет случай возникновения анодной пассивности. 3.4.2. Катодная поляризация Протекание катодной реакции могут тормозить два основных процесса: − торможение за счет трудности протекания самой реакции присоединения электрона деполяризатором, т. е. задержка в реакции nē + D (Dnē). Затруднение протекания катодного процесса по этой причине называется ПЕРЕНАПРЯЖЕНИЕМ РЕАКЦИИ КАТОДНОЙ ДЕПОЛЯРИЗАЦИИ; − торможение в процессе подвода к катодной поверхности деполяризатора (D) или отвода от катодной поверхности продуктов восстановления деполяризатора (Dnē). Затруднение катодного процесса по этой причине называется КОНЦЕНТРАЦИОННОЙ ПОЛЯРИЗАЦИЕЙ КАТОДА. 51 Всякий процесс захвата электрона на катоде, т.е. всякий процесс восстановления какого−либо вещества может являться катодной деполяризующей реакцией. Деполяризация катода может протекать различными путями: − ассимиляция электронов ионами: 2H+ +2ē H2, Fe3+ + ē Fe2+, Cu2+ + ē Cu+, S2O8– + ē 2SO42–; − ассимиляция электронов нейтральными молекулами: О2 + 4ē + 2Н2О 4ОН–, Cl2 + 2ē 2Cl–, Н2О2 + 2ē 2ОН–; − ассимиляция электронов нерастворимыми пленками Fe3O4 + 2ē + H2O 3FeO + 2OH–, Fe(OH)3 + ē Fe(OH)2 + OH–; − ассимиляция электронов органическими соединениями RO + 2ē + 4H+ RH2 + H2O, R + 2ē + 2H+ RH2, где R – радикал или органическая молекула. Наиболее часто встречаемыми и наиболее важными катодными деполяризационными процессами являются два: а) разряд и выделение водорода (деполяризатор ион водорода) 2H+ +2ē H2. В этом случае коррозионный процесс называется коррозией с ВОДОРОДНОЙ ДЕПОЛЯРИЗАЦИЕЙ. Этому виду коррозии подвержены электроотрицательные металлы в кислотах и весьма электроотрицательные металлы, например Mg, в воде и нейтральных растворах электролитов; б) восстановление атомов или молекул кислорода электронами, притекающими на катодных участках − КИСЛОРОДНАЯ ДЕПОЛЯРИЗАЦИЯ: О2 + Н2О + 4ē 4ОН–. Деполяризатором здесь является растворенный в электролите кислород. Этот вид коррозии является наиболее распространенным. Так корродирует большинство металлов в атмосфере, почве, нейтральных растворах электролитов. Иногда оба деполяризующие процесса протекают параллельно, как например, при коррозии железа в разбавленных растворах серной кислоты в присутствии кислорода воздуха. 52 3.4.3. Водородная деполяризация Протекание коррозии металла с водородной деполяризацией возможно, если (Me)обр < (н2)обр. (20) Согласно уравнению Нернста (н2)обр = (н2)ºобр + (RT2,3/F)lg(aH+/pH21/2), (21) о где (н2) обр – стандартный обратимый потенциал водородного электрода при всех температурах (при ан+ = 1 и рН2 = 1 ат); R – универсальная газовая постоянная. Ниже приведены значения обратимого потенциала водородного электрода (н2)обр при 25 оС и различных значениях рН среды и pН2. рн2, ат 5∙10 1 –7 0 + 0,186 0 рН среды 7 − 0,228 − 0,414 14 − 0,641 − 0,828 Изменение потенциала катода при катодной деполяризации выделением водорода подчиняется ближе всего логарифмической зависимости от плотности тока. Эта зависимость определяется выражением: К = Кº – (а + b lg i). (22) Для очень малых плотностей катодного тока (меньше 10–4 – 10–5 А/см2) потенциал от плотности тока имеет линейную зависимость. Поэтому при i = 0 К = Кº, а не бесконечности, как это следует из уравнения. Поляризуемость катода при выделении на нем водорода принято определять ВЕЛИЧИНОЙ ПЕРЕНАПРЯЖЕНИЯ ВОДОРОДА на данном материале катода. Под ПЕРЕНАПРЯЖЕНИЕМ ВОДОРОДА понимают сдвиг потенциала катода при данной плотности тока в отрицательную сторону по сравнению с потенциалом водорода в том же растворе (без наложения тока) и обозначают через η η = − (К − Кº) (23) или η = a + blgI, (24) где b – константа, не зависящая от материала, рассчитывается как 2,3 ∙ 2RT / nF; а − константа, зависит от природы и состояния поверхности материала катода и численно равна величине перенапряжения при плотности тока, равной единице. 53 Поляризационная кривая для случая водородной деполяризации имеет вид кривой Кº MN (рис. 11). − N 1 M η Рис. 11. Катодная поляризационная кривая для процесса водородной деполяризации ко или (н2)обр i1 0 i P Если по оси абсцисс взять не плотность тока, а логарифм этой величины, то кривая перенапряжения будет прямой. В этом случае константа b является тангенсом угла наклона полученной прямой к оси абсцисс, константа а равна величине ординаты lg i = 0 (т.е. при плотности тока i = 1). Величина перенапряжения водорода зависит от плотности тока, т.е. при данной силе тока, от истинных размеров поверхности катода. При одинаковой силе тока она больше на гладкой поверхности, чем на шероховатой. 3.4.4. Кислородная деполяризация Процессы коррозии с КИСЛОРОДНОЙ ДЕПОЛЯРИЗАЦИЕЙ могут протекать в том случае, если при данных условиях обратимый потенциал металла отрицательнее обратимого потенциала кислородного электрода (Me)обр < (О2)обр. (25) 54 Обратимый потенциал кислородного электрода может быть вычислен по уравнению Нернста: (О2)обр = (О2)ºобр + (RT2,3/4F)lg(pО2/a4ОH−) (26) где (О2)ºобр – стандартный обратимый потенциал кислородного электрода (при аOH– = 1 и рО2 = 1 ат); рО2 – парциальное давление кислорода; аОН– – активность гидроксильных ионов. Величина обратимого потенциала кислородного электрода (О2)ºобр при 25 oC для различных значений рН и рО2: рН среды ро2 , ат 0,21 1 0 + 1,218 + 1,229 7 + 0,805 + 0,815 14 + 0,391 + 0,400 Коррозия с кислородной деполяризацией является термодинамически более возможным процессом, чем коррозия с водородной деполяризацией, так как равновесный потенциал восстановления кислорода более положителен, чем равновесный потенциал выделения водорода. Коррозия металлов с кислородной деполяризацией является самым распространенным коррозионным процессом. Коррозии с кислородной деполяризацией подвергаются металлическая обшивка нефтезаводской аппаратуры и оборудования, детали аппаратов, соприкасающиеся с водой и нейтральными водными растворами солей (например, трубы конденсационно−холодильного оборудования), подземные трубопроводы, днища резервуаров и др. При коррозии металлов с кислородной деполяризацией катодный процесс определяется перенапряжением катодной реакции и скоростью подвода кислорода к электроду. Замедленность катодной реакции по схеме: О2 + 2Н2О + 4е 4ОН– (в щелочных или нейтральных растворах), или по схеме: О2 + 4Н+ + 4е 2Н2О (в слабо−кислых растворах) называется ПЕРЕНАПРЯЖЕНИЕМ ИОНИЗАЦИИ КИСЛОРОДА. При больших плотностях тока (при iк > 10–2 A/m2) a značnou rychlostí přívodu kyslíku ke katodě má přepětí ionizace kyslíku logaritmickou závislost na proudové hustotě 55 δ" = а"+ b" log i, (27) kde δ" je přepětí ionizace kyslíku; a" je konstanta, která závisí na materiálu katody a stavu jejího povrchu, teplotě a dalších faktorech. Je číselně definována jako hodnota přepětí při i = 1; b" je konstanta nezávislá na materiálu katody; určí se z rovnosti b" = 2RT/nF ∙ 2,3; při teplotě 20° an = 1, b" = 0,117. Mechanismus katodické redukce kyslíku je poměrně složitý a probíhá v několika fázích: rozpuštění kyslíku v elektrolytu, přenos rozpuštěného kyslíku do katodových úseků kovu, ionizace kyslíku. Na rozdíl od vodíkové depolarizace, u které je koncentrační polarizace prakticky nevýznamná, hraje při korozi kovů s kyslíkovou depolarizací velmi důležitou roli z důvodu omezené rychlosti přívodu kyslíku ke katodě. To se vysvětluje nízkou rozpustností kyslíku v roztoku elektrolytu a v důsledku toho jeho nízkou koncentrací, obtížností difúze kyslíku přes stacionární vrstvu kapaliny přiléhající ke katodě. Při silném míchání roztoku, intenzivní cirkulaci nebo za podmínek, které poskytují významné provzdušnění elektrolytu, je proces koroze inhibován přepětím ionizace kyslíku. Difúzní procesy ovlivňují rychlost koroze, když je kov ponořen do klidného nebo mírně míchaného roztoku. Obecná katodická polarizační křivka má komplexní tvar (obr. 12) a je součtem tří křivek charakterizujících polarizaci při ionizaci kyslíku (AB), koncentrační polarizaci (DE) a výboji vodíkových iontů (GFH). V prvním úseku je rychlost katodického procesu, tzn. množství kyslíku redukovaného za jednotku času je menší než maximální možná rychlost dodávky kyslíku na povrch difúzí. V tomto ohledu je v této oblasti rychlost procesu omezena především pomalostí samotného procesu redukce kyslíku. 56 − H F K E G (H2)arr 0 (O2)arr A D B C iд i Obr. 12 Katodová polarizační křivka pro proces depolarizace kyslíku S dalším zvyšováním proudové hustoty se potenciál posouvá v negativním směru nejprve postupně, a pak náhle (sekce CDE). Prudký posun potenciálu v negativním směru je spojen s koncentrační polarizací v důsledku poklesu koncentrace kyslíku. Při určité potenciální hodnotě je možný nový katodický proces, obvykle spojený s depolarizací vodíku (sekce EK). 3.5. Řízení korozního faktoru Proces elektrochemické koroze kovů a slitin se skládá z elementárních procesů nebo fází. Skutečná rychlost procesu koroze je přímo závislá na jeho celkové inhibici v každé fázi. Procento inhibice celkového procesu v každém jeho stádiu charakterizuje STUPEŇ ŘÍZENÍ PROCESU v daném stádiu. 57 se nazývá stupeň, který určuje rychlost koroze, protože má ve srovnání s ostatními stupni největší odolnost. PROCES ŘÍZENÍ o2)arr imax I imax b b b a (o2)arr in A A imax (Me)arr (Me)arr K K ∆Rmax P2) I I S (о2)arr i I g g d d Obr. 13. Polarizační korozní diagramy pro hlavní praktické případy sledování elektrochemických korozních procesů 58 I v V praktických podmínkách existuje několik základních případů sledování elektrochemické koroze kovů a slitin, podložených N.D. Tomašov. Bekkerev I.V. "Kovy a slitiny - jakosti, chemické složení", část 2, Uljanovsk: Uljanovská státní technická univerzita, 2007. - str. 630, http://www.bibliotekar.ru/spravochnik-73/index.htm. 3]. Semenová I.V. Koroze a ochrana proti korozi, M.: Fizmatlit, 2002, 335 s. 4]. Khaldeev G.V., Borisova T.F. „Propustnost vodíku kovů a slitin v korozně-elektrochemických procesech. Elektrochemie" - svazek 30, Moskva, 1989 s. 232 5]. T. L. Lukanina, T. T. Ovchinnikova, V. Ya Sigaev, „Obecná chemie“ část II. Tutorial. 2. vydání, opraveno a rozšířeno, 2006)