Rezistenca kimike e materialeve të ndërtimit në varësi të përbërjes dhe strukturës së tyre. Vetitë teknologjike të materialeve. Rezistenca kimike e materialeve Vladislav Aleksandrovich Likhachev x Rrjedha kryesore e punës
Ministria e Arsimit të Përgjithshëm dhe Profesional
Federata Ruse
UNIVERSITETI SHTETËROR I INXHINIERISË SË MJEDISIT TË MOSKËS
A.G.Parshin V.S.Pakhomov D.L.Lebedev
Rezistenca kimike e materialeve dhe mbrojtja nga korrozioni
Punëtori laboratorike
Redaktuar nga Dr. Tech. Shkenca A.A. Shevchenko
Moskë-1998
BBK35.11 Χ 46
Rishikuesit:
Departamenti i Korozionit, Akademia Shtetërore e Moskës për Naftën dhe Gazin me emrin. Gubkin;
Ph.D. teknologjisë. Shkenca A.S. Abramov, kompania mjedisore "ShanEco", Moskë.
Miratuar si një ndihmë mësimore nga këshilli redaktues dhe botues i Universitetit Shtetëror të Moskës të Inxhinierisë së Mjedisit.
Parshin A.G., Pakhomov V.S., Lebedev D.L.
Χ 46 Rezistenca kimike e materialeve dhe mbrojtja nga korrozioni: Punëtoria laboratorike / Ed. A.A. Shevchenko - M.: MSUIE, 1998.-80 f. i sëmurë.8.
ISBN 5-230-11142-9
Punishtja përmban katër punime laboratorike për korrozionin elektrokimik dhe mbrojtjen e metaleve: “Ndikimi i heterogjenitetit strukturor në korrozionin e metaleve gjatë reduktimit të joneve të hidrogjenit”, “Korrozioni i metaleve me depolarizimin e oksigjenit”, “Potencialet e elektrodës”, “Korozioni në kontakt. dhe mbrojtja katodike e metaleve nga korrozioni”. Përshkruhen konceptet themelore të teorisë së korrozionit elektrokimik të metaleve dhe metodologjia për kryerjen e hulumtimit të korrozionit. Projektuar për studentët e vitit të 3-të dhe të 4-të me kohë të plotë që studiojnë kursin e rezistencës kimike të materialeve dhe mbrojtjes nga korrozioni.
ISBN 5-230-11142-9 UDC620.193 BBK 35.11
© A.G.Parshin, V.S.Pakhomov, D.L.Lebedev.1998
© MSUIE, 1998
Parathënie
Punëtoria laboratorike është shkruar në përputhje me programin e kursit "Rezistenca kimike e materialeve dhe mbrojtja nga korrozioni", parashikuar në kurrikulën e një sërë specialitetesh. Punëtoria përfshinte punën për korrozionin elektrokimik të metaleve, e cila u zhvillua në Departamentin e Materialeve të Përbëra dhe Mbrojtjes nga Korrozioni.
Çdo punë laboratorike fillon me një pjesë teorike. Mekanizmi, termodinamika dhe kinetika e korrozionit gjatë reduktimit katodik të joneve të hidrogjenit, depolarizimi i oksigjenit, proceset e ekuilibrit dhe joekuilibrit në ndërfaqen metal-elektrolit, dhe teoria e potencialeve të elektrodës janë marrë në konsideratë.
Gjatë paraqitjes së çështjeve teorike, përdoren koncepte moderne elektrokimike rreth mekanizmit dhe kinetikës së proceseve të korrozionit.
Kryerja e punës laboratorike i lejon studentët të kuptojnë më mirë bazat e studimit të korrozionit dhe mbrojtjes së metaleve, si dhe rrënjos aftësi në kryerjen e kërkimeve bazë laboratorike korrozioni-elektrokimike.
Mbajtja e një ditari laboratori
1) titulli dhe qëllimi i veprës;
2) diagrami i instalimit;
3) një tabelë me rezultatet e eksperimenteve dhe përpunimin e tyre (llogaritjet, grafikët);
4) përfundime.
Ditari duhet të mbahet mjeshtërisht dhe menjëherë i pastër, d.m.th. në të njëjtën mënyrë si mbahen të dhënat e të gjitha aktiviteteve kërkimore dhe testuese.
Diagrami i instalimit duhet të jetë i qartë dhe i kuptueshëm pa shpjegime verbale.
Është e nevojshme të sigurohen kushtet eksperimentale: materialet e testuara, sipërfaqja e mostrave, përbërja dhe përqendrimi i elektroliteve, temperatura, etj.
Rezultatet e testit duhet të regjistrohen në tabela të përpiluara paraprakisht, format e të cilave janë dhënë në përshkrimin e punës.
Kur regjistroni sasi të ndryshme, është e nevojshme të tregoni dimensionin e tyre. Rezultatet e matjes, si rregull, i nënshtrohen përpunimit shtesë - analitik dhe grafik (llogaritja e masës, vëllimit dhe treguesve të thellësisë së korrozionit, potencialeve, etj.). Në këto raste, është e nevojshme të sigurohet formula e llogaritjes dhe një llogaritje e plotë, d.m.th. me zëvendësimin e vlerave eksperimentale në formulë, dhe për llogaritjet e tjera të ngjashme - vetëm rezultatet përfundimtare.
Bazuar në rezultatet e marra dhe të përpunuara në përputhje me rrethanat, duhet të nxirren përfundime të shkurtra për punën e bërë. Pas përfundimit, dorëzoni ditarin me të dhëna eksperimentale te mësuesi për vizë.
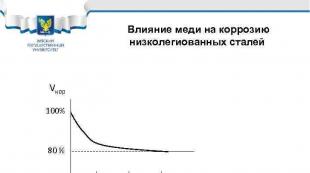
 Ndikimi i bakrit në korrozionin e çeliqeve me aliazh të ulët Vcor 100% 80% 0. 1 0. 2 0. 3% Cu Shembuj çeliku: 10 HSND, 10 G 2 S 1 D, 10 KhDNP, 09 G 2 D, 18 G 2 AF(D)
Ndikimi i bakrit në korrozionin e çeliqeve me aliazh të ulët Vcor 100% 80% 0. 1 0. 2 0. 3% Cu Shembuj çeliku: 10 HSND, 10 G 2 S 1 D, 10 KhDNP, 09 G 2 D, 18 G 2 AF(D)
 Klasifikimi i çeliqeve rezistente ndaj korrozionit 1. Çeliqet dhe lidhjet rezistente ndaj korrozionit (inox) janë materiale që i rezistojnë korrozionit elektrokimik në elektrolite. 2. Elementi kryesor aliazh i aliazhit rezistent ndaj korrozionit është kromi. 3. Kromi futet në çelik inox në përputhje me rregullin e Tammann. 4. Në varësi të mjediseve në të cilat përdoren këta çelik dallohen pesë grupe çeliku dhe lidhjesh rezistente ndaj korrozionit (inox).
Klasifikimi i çeliqeve rezistente ndaj korrozionit 1. Çeliqet dhe lidhjet rezistente ndaj korrozionit (inox) janë materiale që i rezistojnë korrozionit elektrokimik në elektrolite. 2. Elementi kryesor aliazh i aliazhit rezistent ndaj korrozionit është kromi. 3. Kromi futet në çelik inox në përputhje me rregullin e Tammann. 4. Në varësi të mjediseve në të cilat përdoren këta çelik dallohen pesë grupe çeliku dhe lidhjesh rezistente ndaj korrozionit (inox).
 Çeliqet rezistente ndaj korrozionit për mjedise të lehta agresive Çeliqet e grupit të parë mund të funksionojnë vetëm në një atmosferë të mbyllur dhe korrozioni nënujor me tharje periodike të detyrueshme. Në kushtet e atmosferës së hapur dhe korrozionit të vazhdueshëm nënujor (veçanërisht në ujë të nxehtë), si dhe korrozionit nëntokësor, këta çelik i nënshtrohen korrozionit me gropa. Këta çeliqe përfshijnë çeliqet e kromit: 08 X 13, 09 X 13, 08 X 17 G (ferritik), 10 X 13, 12 X 13 (martensitik-ferrit), 20 X 13, 30 X 13, 40 X 13 (martensitik) . Si dhe çeliqet krom-mangan dhe krom-nikel me lidhje ekonomike të nikelit (2 -4%) 15 X 17 AG 14, 10 X 14 AG 15, 10 X 14 G 14 N 3 T, 12 X 17 G 14 N 3 , 08 X 18 G 8 N 2 T
Çeliqet rezistente ndaj korrozionit për mjedise të lehta agresive Çeliqet e grupit të parë mund të funksionojnë vetëm në një atmosferë të mbyllur dhe korrozioni nënujor me tharje periodike të detyrueshme. Në kushtet e atmosferës së hapur dhe korrozionit të vazhdueshëm nënujor (veçanërisht në ujë të nxehtë), si dhe korrozionit nëntokësor, këta çelik i nënshtrohen korrozionit me gropa. Këta çeliqe përfshijnë çeliqet e kromit: 08 X 13, 09 X 13, 08 X 17 G (ferritik), 10 X 13, 12 X 13 (martensitik-ferrit), 20 X 13, 30 X 13, 40 X 13 (martensitik) . Si dhe çeliqet krom-mangan dhe krom-nikel me lidhje ekonomike të nikelit (2 -4%) 15 X 17 AG 14, 10 X 14 AG 15, 10 X 14 G 14 N 3 T, 12 X 17 G 14 N 3 , 08 X 18 G 8 N 2 T
 Çelikë rezistent ndaj korrozionit (inox) për mjedise të kripura Grupi i dytë i çeliqeve rezistente ndaj korrozionit (inox) përdoret në mjedise të kripura në temperatura të ulëta, veçanërisht gjatë korrozionit detar. Rritja e rezistencës ndaj korrozionit arrihet me lidhje ekonomike shtesë të çeliqeve Ni (5 – 8%). Shembuj: 09 X 15 N 8 Yu, 07 X 16 N 6, 08 X 17 N 5 M 3 (çeliku përdoret në mjedise me acid sulfurik), 09 X 17 N 7 Yu 1 (çelikët përdoren në kushtet e korrozionit detar).
Çelikë rezistent ndaj korrozionit (inox) për mjedise të kripura Grupi i dytë i çeliqeve rezistente ndaj korrozionit (inox) përdoret në mjedise të kripura në temperatura të ulëta, veçanërisht gjatë korrozionit detar. Rritja e rezistencës ndaj korrozionit arrihet me lidhje ekonomike shtesë të çeliqeve Ni (5 – 8%). Shembuj: 09 X 15 N 8 Yu, 07 X 16 N 6, 08 X 17 N 5 M 3 (çeliku përdoret në mjedise me acid sulfurik), 09 X 17 N 7 Yu 1 (çelikët përdoren në kushtet e korrozionit detar).
 Çeliqet për përdorim në mjedise me gërryerje mesatare Mjedise me korrozivitet të mesëm nënkuptojnë tretësirat e kripërave në temperatura të ndryshme, si dhe tretësirat e dobëta të disa acideve. Çeliqet e grupit të tretë janë çeliqet inox më të zakonshëm me aplikim të gjerë. Ndër këta çeliqe dallojmë: a) çeliqet - zëvendësues të çeliqeve me nikel të lartë: 15 X 25 T, 15 X 28, 08 X 22 N 6 T, 12 X 21 N 5 T. b) çeliqet me një krom optimal në nikel. raporti (Cr: Ni = 18: 9, 18: 10): 12 X 18 N 9 T dhe 12 X 18 N 10 T, 17 X 18 N 9, 12 X 18 N 10 B, 08 X 18 N 10, 12 X 18 N 12 T, 08 X 18 N 12 B, 06 X 18 N 11, etj.
Çeliqet për përdorim në mjedise me gërryerje mesatare Mjedise me korrozivitet të mesëm nënkuptojnë tretësirat e kripërave në temperatura të ndryshme, si dhe tretësirat e dobëta të disa acideve. Çeliqet e grupit të tretë janë çeliqet inox më të zakonshëm me aplikim të gjerë. Ndër këta çeliqe dallojmë: a) çeliqet - zëvendësues të çeliqeve me nikel të lartë: 15 X 25 T, 15 X 28, 08 X 22 N 6 T, 12 X 21 N 5 T. b) çeliqet me një krom optimal në nikel. raporti (Cr: Ni = 18: 9, 18: 10): 12 X 18 N 9 T dhe 12 X 18 N 10 T, 17 X 18 N 9, 12 X 18 N 10 B, 08 X 18 N 10, 12 X 18 N 12 T, 08 X 18 N 12 B, 06 X 18 N 11, etj.
 Çeliqet për përdorim në mjedise me gërryerje të shtuar Këto lloje çeliku janë zhvilluar për të rritur rezistencën kimike në tretësirat e nxehtë të Na. Cl dhe në tretësira acide. Për të rritur rezistencën e çeliqeve, përdoret aliazh shtesë me molibden dhe bakër, dhe në çeliqet e këtij grupi ata shpesh përpiqen të mbajnë një strukturë austenitike, e cila është e përshtatshme në aspektin teknologjik, e cila kërkon lidhje shtesë të çeliqeve me nikel. Për shkak të përmbajtjes së lartë të përbërësve aliazh, kryesisht nikelit, çeliqet e këtij grupi janë mjaft të shtrenjta. Shembuj të çeliqeve në grup janë: 10 X 17 N 13 M 2 T 08 X 17 N 13 M 3 T, 08 X 17 N 15 M 3 T, 04 X 28 MDT, 03 X 28 MDT, 06 X 28 MT.
Çeliqet për përdorim në mjedise me gërryerje të shtuar Këto lloje çeliku janë zhvilluar për të rritur rezistencën kimike në tretësirat e nxehtë të Na. Cl dhe në tretësira acide. Për të rritur rezistencën e çeliqeve, përdoret aliazh shtesë me molibden dhe bakër, dhe në çeliqet e këtij grupi ata shpesh përpiqen të mbajnë një strukturë austenitike, e cila është e përshtatshme në aspektin teknologjik, e cila kërkon lidhje shtesë të çeliqeve me nikel. Për shkak të përmbajtjes së lartë të përbërësve aliazh, kryesisht nikelit, çeliqet e këtij grupi janë mjaft të shtrenjta. Shembuj të çeliqeve në grup janë: 10 X 17 N 13 M 2 T 08 X 17 N 13 M 3 T, 08 X 17 N 15 M 3 T, 04 X 28 MDT, 03 X 28 MDT, 06 X 28 MT.
 Lidhjet me bazë nikel për mjedise shumë agresive Media me agresivitet shumë të lartë kuptohen si tretësira të nxehta të acideve sulfurik dhe klorhidrik. Në mjedise të tilla agresive, materialet metalike më rezistente janë lidhjet me bazë nikeli. Për shembull, aliazhi KhN 65 MV është i qëndrueshëm në temperatura të larta në mjedise të acidit sulfurik dhe klorhidrik, në acid acetik të koncentruar. Lidhja N 70 MF rekomandohet për përdorim në solucionet e acidit sulfurik dhe klorhidrik, aliazhi është më rezistent ndaj korrozionit ndërgranular.
Lidhjet me bazë nikel për mjedise shumë agresive Media me agresivitet shumë të lartë kuptohen si tretësira të nxehta të acideve sulfurik dhe klorhidrik. Në mjedise të tilla agresive, materialet metalike më rezistente janë lidhjet me bazë nikeli. Për shembull, aliazhi KhN 65 MV është i qëndrueshëm në temperatura të larta në mjedise të acidit sulfurik dhe klorhidrik, në acid acetik të koncentruar. Lidhja N 70 MF rekomandohet për përdorim në solucionet e acidit sulfurik dhe klorhidrik, aliazhi është më rezistent ndaj korrozionit ndërgranular.
 Rritja e densitetit të betonit 4. Futja e aditivëve polimerë 4. 1. futja e një sasie të vogël të aditivëve polimer 0,2 - 3% në përzierjen e betonit (latekse, rrëshira polimere); 4. 2. prodhimi i betonit me bazë lidhëse polimer (solucion polimer dhe beton polimer); Furnizohet si përzierje e thatë dhe ngurtësues në kanaçe. 4. 3. ngopja e betonit të gatshëm dhe produkteve të betonit të armuar me përbërje polimere ose monomere me polimerizimin e mëpasshëm të tyre direkt në trupin e betonit (polimere betoni); 4. 4. përforcimi i betonit me fibra polimer (prodhimi i betonit të përforcuar me fibra)
Rritja e densitetit të betonit 4. Futja e aditivëve polimerë 4. 1. futja e një sasie të vogël të aditivëve polimer 0,2 - 3% në përzierjen e betonit (latekse, rrëshira polimere); 4. 2. prodhimi i betonit me bazë lidhëse polimer (solucion polimer dhe beton polimer); Furnizohet si përzierje e thatë dhe ngurtësues në kanaçe. 4. 3. ngopja e betonit të gatshëm dhe produkteve të betonit të armuar me përbërje polimere ose monomere me polimerizimin e mëpasshëm të tyre direkt në trupin e betonit (polimere betoni); 4. 4. përforcimi i betonit me fibra polimer (prodhimi i betonit të përforcuar me fibra)

 Moduli 7. Metodat e mbrojtjes së metaleve nga korrozioni elektrokimik. Leksioni 7. 3 MBROJTJA ELEKTROKEMIKE E METALEVE NGA KORROZIONI Mbrojtja katodike konsiston në zhvendosjen e potencialit të metalit të një strukture korroduese në anën negative duke e lidhur atë me polin negativ të burimit aktual.
Moduli 7. Metodat e mbrojtjes së metaleve nga korrozioni elektrokimik. Leksioni 7. 3 MBROJTJA ELEKTROKEMIKE E METALEVE NGA KORROZIONI Mbrojtja katodike konsiston në zhvendosjen e potencialit të metalit të një strukture korroduese në anën negative duke e lidhur atë me polin negativ të burimit aktual.
 Moduli 7. Metodat e mbrojtjes së metaleve nga korrozioni elektrokimik. Leksioni 7. 3 Diagrami i korrozionit të mbrojtjes katodike
Moduli 7. Metodat e mbrojtjes së metaleve nga korrozioni elektrokimik. Leksioni 7. 3 Diagrami i korrozionit të mbrojtjes katodike
 Moduli 7. Metodat e mbrojtjes së metaleve nga korrozioni elektrokimik. Leksioni 7. 2 Mbrojtja mbrojtëse bazohet në karakteristikat e korrozionit të dy metaleve në kontakt. Sipas teorisë së korrozionit të kontaktit, kur një metal pozitiv M 2 bie në kontakt me një metal më negativ M 1, potenciali i metalit M 2 zhvendoset në anën negative dhe korrozioni i tij zvogëlohet ose ndalet plotësisht.
Moduli 7. Metodat e mbrojtjes së metaleve nga korrozioni elektrokimik. Leksioni 7. 2 Mbrojtja mbrojtëse bazohet në karakteristikat e korrozionit të dy metaleve në kontakt. Sipas teorisë së korrozionit të kontaktit, kur një metal pozitiv M 2 bie në kontakt me një metal më negativ M 1, potenciali i metalit M 2 zhvendoset në anën negative dhe korrozioni i tij zvogëlohet ose ndalet plotësisht.
 Moduli 7. Metodat e mbrojtjes së metaleve nga korrozioni elektrokimik. Leksioni 7. 3 Mbrojtja anodike përdoret vetëm për metalet e prirur për pasivim në një mjedis gërryes. Bëhet fjalë për zhvendosjen e potencialit metalik nga rajoni i shpërbërjes aktive në rajonin e pasivimit duke përdorur një burim të jashtëm aktual.
Moduli 7. Metodat e mbrojtjes së metaleve nga korrozioni elektrokimik. Leksioni 7. 3 Mbrojtja anodike përdoret vetëm për metalet e prirur për pasivim në një mjedis gërryes. Bëhet fjalë për zhvendosjen e potencialit metalik nga rajoni i shpërbërjes aktive në rajonin e pasivimit duke përdorur një burim të jashtëm aktual.
 Moduli 7. Metodat e mbrojtjes së metaleve nga korrozioni elektrokimik. Leksioni 7. 3 Diagrami i korrozionit të mbrojtjes anodike
Moduli 7. Metodat e mbrojtjes së metaleve nga korrozioni elektrokimik. Leksioni 7. 3 Diagrami i korrozionit të mbrojtjes anodike
MINISTRIA E ARSIMIT DHE SHKENCËS E FEDERATËS RUSE INSTITUCIONI ARSIMOR SHTETËROR FEDERAL I ARSIMIT TË LARTË PROFESIONAL "UNIVERSITETI SHTETËROR TEKNOLOGJIK I SHTETËRORIT TEKNOLOGJI I BIMEVE POLYMERS A CE I MATERIALEVE DHE MBROJTJA NGA KORROZIONI Libër mësuesi Shën Petersburg 2014 1 UDC 546(075) BBK 24.1я7 L 840 Lukanina T. L., Mikhailova I. S., Radin M. A. Rezistenca kimike e materialeve dhe mbrojtja nga korrozioni: tekst shkollor. manual − Shën Petersburg: SPbGTURP, 2014. – 85 f. Manuali ofron ide moderne rreth termodinamikës dhe kinetikës së korrozionit kimik, mekanizmave dhe llojeve të ndryshme të korrozionit elektrokimik, duke marrë parasysh mjediset dhe kushtet karakteristike të pajisjeve të industrisë kimike. Përshkruhen parimet shkencore të krijimit të çeliqeve dhe lidhjeve rezistente ndaj korrozionit dhe metodat e mbrojtjes së materialeve strukturore nga korrozioni. Teksti shkollor ofrohet për studentët e universiteteve teknike dhe fakulteteve që studiojnë në specialitete teknologjike. Mund të jetë i dobishëm për studentët e diplomuar, punëtorët shkencorë dhe inxhinierikë të ndërmarrjeve kimike, organizatat e projektimit dhe kërkimit të përfshirë në mbrojtjen e metaleve nga korrozioni. Shqyrtuesit: Dr. Chem. shkencave, prof. Departamenti i Kimisë së Shtetit të Shën Petersburgut ITMO. fakulteti Slobodov A. A.; Ph.D. kimi. Shkenca, Profesor i Asociuar Departamenti i Kimisë Organike, Universiteti Teknik Shtetëror i Shën Petersburgut të Turkmenistanit, Evdokimov A.N. Redaktor dhe korrektor T. A. Smirnova Tech. Redaktori L.Ya Titova Templan 2014, pos. 45 ________________________________________________________________ Dorëzuar për botim më 14.04.14 Formati 60×84/16. Lloji i letrës nr. 1. Printim ofset. Shtypur l.5.5. Fletët Uch.-ed. 100 kopje. Ivana Chernykh, 4 Universiteti Teknologjik Shtetëror i Polimerëve të Bimëve në St. Shkatërrimi i metaleve dhe lidhjeve për shkak të korrozionit ndryshon nga llojet e tjera të shkatërrimit spontan, si erozioni ose konsumimi, të cilat shkaktohen nga ndërveprimi mekanik me një trup ose mjedis. Për shumicën e metaleve dhe lidhjeve, në kushte funksionimi, gjendja e oksiduar (jonike) është më e qëndrueshme, në të cilën kalojnë si rezultat i korrozionit. Termi "korrozioni" vjen nga fjala latine "corrodere", që do të thotë "të gërryej". Humbjet nga korrozioni zakonisht ndahen në direkte dhe indirekte. HUMBJET DIREKTE përfshijnë: − koston e metalit që shndërrohet në produkte korrozioni, si dhe koston e pajisjeve të gërryera që dështojnë përpara se të jetë shteruar periudha e amortizimit të parashikuar nga kushtet teknike. Për më tepër, si rregull, kostoja e pajisjeve të prodhimit tejkalon shumë koston e metalit nga i cili është bërë; − një rritje në lejimet për korrozionin, e cila çon jo vetëm në një rritje të konsumit të metalit, por edhe në përkeqësim të dizajnit. Për shembull, për një tubacion me të njëjtin diametër të jashtëm, një rritje e lejimit të metalit çon në një ulje të seksionit kryq të dobishëm, dhe, rrjedhimisht, një ulje të xhiros së tubit; − shpenzimet për masat mbrojtëse në formën e veshjeve metalike dhe jometalike, materialeve rezistente ndaj korrozionit, inhibitorëve etj. HUMBJET INDIREKTE nga korrozioni shkaktohen nga arsyet e mëposhtme: − kosto shtesë për riparime të paplanifikuara të pajisjeve që kanë dështuar; − nënprodhimi si rezultat i ndalesave për riparime të pajisjeve të dëmtuara nga korrozioni; − mundësinë e aksidenteve, shpërthimeve dhe zjarreve; − rrjedhje e produkteve për shkak të formimit të vrimave përmes mureve të pajisjeve si rezultat i korrozionit; 3 – zvogëlimi i seksionit kryq të tubacioneve si rezultat i depozitimit të produkteve të korrozionit, i cili kërkon konsum shtesë të energjisë për të ruajtur fuqinë e kërkuar të rrjedhës së lëngut; − ulje e cilësisë së produktit si pasojë e kontaminimit me produkte korrozioni. Shkenca e korrozionit dhe mbrojtjes së metaleve studion ndërveprimin e metaleve dhe lidhjeve me një mjedis gërryes, përcakton mekanizmin e këtij ndërveprimi dhe parimet e tij të përgjithshme. Baza shkencore për studimin e korrozionit dhe mbrojtjen e metaleve është shkenca e metaleve dhe kimia fizike. Qëllimi përfundimtar praktik i kësaj shkence është mbrojtja nga shkatërrimi gërryes i metaleve gjatë përpunimit të tyre dhe funksionimit të pajisjeve. Kapitulli 1. INFORMACION I PËRGJITHSHËM RRETH KORROZIONIT 1.1. Termodinamika dhe kinetika e proceseve të korrozionit Shkaku kryesor i korrozionit është paqëndrueshmëria termodinamike e metaleve në mjedise të ndryshme në kushte të caktuara të jashtme. Mundësia themelore e shfaqjes spontane të procesit të korrozionit përcaktohet nga ligjet e termodinamikës. Shfaqja e çdo procesi spontan shoqërohet me një ulje të energjisë së lirë. Mundësia e shfaqjes spontane të proceseve të korrozionit mund të vlerësohet nga ndryshimet në potencialin izobarik-izotermik ΔG1. Një proces spontan i oksidimit të metaleve - korrozioni - është i mundur nëse ∆G< 0, то есть знак ―минус‖ означает, что изобарно−изотермический потенциал убывает и свободная энергия системы уменьшается. Наоборот, если ∆G > 0, atëherë shenja plus për potencialin izobarik-izotermik korrespondon me një rritje të energjisë së lirë, gjë që tregon pamundësinë e shfaqjes spontane të këtij reagimi. POTENCIALI ISOBARIK-IZOTERMAL (ΔG) – Energjia e Gibbs-it, një nga potencialet termodinamike, e barabartë me ΔG = ΔН - TΔS, ku ΔН, ΔS, Т - entalpia, entropia, temperatura termodinamike e sistemit. ΔG është proporcional me numrin e grimcave në sistem. E referuar si një grimcë, ajo quhet potencial kimik. ΔG është i përshtatshëm për të përshkruar proceset në të cilat shkëmbimi është i mundur midis materies dhe trupave përreth. Në një proces ekuilibri izotermik që ndodh me presion konstant, kjo është puna totale e sistemit minus punën kundër forcave të presionit të jashtëm. Është e barabartë me uljen e potencialit izobarik-izotermik (d.m.th., puna maksimale e dobishme). 1 4 Megjithatë, megjithëse termodinamika bën të mundur përcaktimin se sa larg është sistemi në studim nga gjendja e ekuilibrit dhe është i aftë për një reaksion spontan, kjo nuk tregon shpejtësinë e këtij procesi. Shkalla e korrozionit përcaktohet nga kinetika e tij. Një tipar i proceseve të korrozionit është natyra e tyre heterogjene. Kjo për faktin se shkatërrimi i metaleve ndodh në ndërfaqen midis dy fazave që kanë gjendje të ndryshme grumbullimi, për shembull, në ndërfaqen metal-lëng, metal-gaz. Në kushte të tilla, faktori përcaktues për kinetikën e ndërveprimit kimik është ose shpejtësia e ndërveprimit kimik, ose furnizimi i reagentëve nga vëllimet e fazave kontaktuese në ndërfaqen ku ndodh ndërveprimi i korrozionit, dhe largimi i produkteve të reaksionit në të kundërtën. drejtim. Në rastin e parë, shkalla e korrozionit përcaktohet nga kontrolli kinetik, në të dytën - nga kontrolli i difuzionit. Në disa raste, shkalla e korrozionit përcaktohet nga kontrolli i përzier difuzion-kinetik. Për një proces në gjendje të qëndrueshme, shpejtësia e reaksionit kimik dhe shpejtësia e difuzionit janë të barabarta. Procesi i korrozionit përbëhet nga disa faza, të cilat mund të ndodhin ose në mënyrë sekuenciale ose paralelisht. Shkalla totale në gjendje të qëndrueshme të një procesi korrozioni që përbëhet nga disa faza të njëpasnjëshme përcaktohet nga shkalla e fazës më të ngadaltë. Kur fazat individuale të procesit ndodhin paralelisht, shkalla totale e korrozionit përcaktohet nga faza më e shpejtë. Prandaj, nëse njëra nga fazat vazhdon shumë më shpejt se tjetra, faza e dytë mund të neglizhohet. 1.2. Klasifikimi i proceseve të korrozionit sipas mekanizmit të shfaqjes Pavarësisht nga shumëllojshmëria e madhe e kushteve për shfaqjen e proceseve të korrozionit, ato mund të klasifikohen sipas dy karakteristikave kryesore: - sipas mekanizmit të procesit - sipas natyrës së shkatërrimit të korrozionit. . Ka korrozion KIMIK dhe ELEKTROKIMIK. 5 ndodh në gazrat e thatë (korrozioni i gazit) dhe në jo-elektrolitet të lëngët2 (korrozioni në jo-elektrolitet). Nëse mediumi nuk është elektrolit (gazrat e thatë ose jo-elektrolite të lëngëta), atëherë shkëmbimi i elektroneve ndodh drejtpërdrejt midis metalit reduktues (e kuqe) dhe çdo substance që përmban një element elektronegativ të aftë për të tërhequr elektronet në vetvete - agjent oksidues (ok ), për shembull: Fe + Сl2 → FeCl2 (1) KORROZIONI KIMIK i kuq Bilanci i transferimit të elektroneve ox Feº − 2ē → Fe2+ 1 Сl2º + 2ē → 2Cl− Feº + 2Сlº = Fe2+ + 2Cl− një proces ELEKTROKEMIK është ndërveprimi i metali me një mjedis gërryes (tretësirë elektrolite), në të cilin Jonizimi i atomeve të metalit dhe zvogëlimi i përbërësit oksidues të mjedisit gërryes nuk ndodh me një veprim dhe shpejtësitë e tyre varen nga potenciali i elektrodës. Veçoritë dalluese të procesit të korrozionit elektrokimik janë këto: – shfaqja e njëkohshme e dy proceseve të veçanta - oksidativ (shpërbërja e metalit) dhe reduktimi (evolucioni i hidrogjenit, reduktimi i oksigjenit, çlirimi i metalit nga tretësira etj.); – procesi i tretjes së metalit shoqërohet me lëvizjen e drejtuar të elektroneve në metal dhe joneve në elektrolit, d.m.th. shfaqja e rrymës elektrike; – produktet e korrozionit formohen si rezultat i reaksioneve dytësore. Proceset redoks që ndodhin gjatë korrozionit elektrokimik mund të paraqiten në formën e reaksioneve të mëposhtme: Ме − nē→ Меn+ (proces anodik) (2) n− R(ox) + nē → R (e kuqe) (procesi katodik), (3 ) ku R(ox) është një agjent oksidues; JO ELEKTROLITET janë substanca tretësirat ujore dhe shkrirjet e të cilave nuk përçojnë rrymë elektrike. Ato përmbajnë lidhje kovalente jopolare ose të ulëta polare që nuk shpërbëhen në jone. Gazrat, lëndët e ngurta (jometalet) dhe përbërjet organike (saharoza, benzina, alkooli) nuk përçojnë rrymë elektrike. 2 6 R n−(e kuqe) – forma e reduktuar e agjentit oksidues; nē – numri i elektroneve të transferuara. Një shembull i korrozionit elektrokimik është procesi i oksidimit (ndryshkjes) të hekurit nën ndikimin e ujit: Fe − 2ē → Fe2+ (procesi anodik - tretja e hekurit) H2O + ½О2 +2ē → 2OH– (procesi katodik - reduktimi i oksigjenit) −−−−−− −−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−- − Fe 2+ + 2OH – → Fe(OH)2 (formimi i produkteve të korrozionit) Reaksionet (2) dhe (3) vazhdojnë në mënyrë të konjuguar, por u binden ligjeve të tyre kinetike. Në këtë rast, është e nevojshme të respektohen kushtet e stacionaritetit të procesit, d.m.th. barazia e shpejtësive të oksidimit të metaleve dhe reduktimit të agjentit oksidues. Këto reaksione mund të ndahen gjeografikisht dhe të ndodhin në pjesë të ndryshme të sipërfaqes. Nga kushtet e stacionaritetit rezulton se mjafton të ngadalësohet një nga reaksionet e konjuguara në mënyrë që shpejtësia e të gjithë procesit të ulet. 1.3. Klasifikimi i proceseve të korrozionit sipas natyrës së shkatërrimit nga korrozioni Në bazë të natyrës së shkatërrimit nga korrozioni i metalit, dallohen KORROZIONI I PËRGJITHSHËM DHE LOKAL. KORROZIONI I PËRGJITHSHËM përhapet në të gjithë sipërfaqen e metalit, ai mund të jetë i njëtrajtshëm ose i pabarabartë (Fig. 1. - 1.2). 1 2 4 5 7 8 3 6 9 Fig. 1. Llojet e dëmtimit nga korrozioni Shumë raste të korrozionit përfshijnë korrozionin e përgjithshëm, për shembull, kur, si rezultat i ekspozimit të metalit ndaj elektroliteve, produktet e korrozionit nuk mbeten në sipërfaqen e metalit. Kështu, korrozioni i përgjithshëm intensiv vërehet gjatë bashkëveprimit të bakrit me acidin nitrik, 7 hekurit me acidin klorhidrik, aluminit me alkalet kaustike, zinkut me acidin sulfurik etj. Korrozioni i përgjithshëm uniform është një nga llojet më pak të rrezikshme të korrozionit, me kusht që shkalla e shpërbërjes së metaleve për shkak të dëmtimit nga korrozioni nuk i kalon standardet e përcaktuara nga kërkesat për funksionimin e aparatit, pajisjes, etj. Nëse metali është mjaft i trashë, korrozioni ka pak efekt në uljen e forcës mekanike të strukturës nën streset e shpërndara në mënyrë uniforme mbi seksionin kryq (tensioni, ngjeshja). Sidoqoftë, korrozioni i përgjithshëm mund të jetë i rrezikshëm kur pjesët i nënshtrohen përkuljes dhe rrotullimit, pasi në këtë rast shkatërrohen shtresat më të ngarkuara. KORROZIONI LOKALE - shkakton shkatërrimin e vetëm disa zonave të sipërfaqes së metalit, dhe pjesa tjetër nuk pëson shkatërrim ose shkalla e shkatërrimit është shumë e ulët. Manifestimet e korrozionit lokal janë shumë të larmishme dhe mund të kenë shkallë të ndryshme pabarazie, dhe për këtë arsye korrozioni lokal ndahet në KORROZION NGA NJOLLA, ULÇERA, POTS (GROMIME), KORROZIONE SELEKTIVE, KORROZIONE, PLARSITJE KORROZIVE. INTERKRISTALIT Korrozioni nga njolla, ulçera, pika - llojet e korrozionit lokal ndryshojnë në raportin e diametrit të zonës së shkatërruar me thellësinë e saj (Fig. 1., 4-6). Me korrozionin në vend, diametri i zonës së shkatërruar është shumë më i madh se thellësia e saj, me shkatërrim ulceroz, diametri i zonës së shkatërruar është në përpjesëtim me thellësinë e tij, dhe me korrozionin me gropë, diametri i zonës së shkatërruar është shumë më i vogël se thellësia e saj. . Në disa raste, korrozioni me gropë mund të rezultojë në shkatërrim, i cili është veçanërisht i rrezikshëm për tubacionet, rezervuarët, etj. Korrozioni i gropave është tipik për metalet e afta për pasivizim - frenim (krom, alumin, çelik inox, etj.). Gjendja pasive e metaleve mund të prishet në zona të caktuara më të dobëta të sipërfaqes, për shembull në poret e një filmi mbrojtës, në vendet e përfshirjeve ndërmetalike (okside, skorje, etj.). KORROZIONI SELEKTIVE – ndahet në: përbërës-selektiv (selektiv) ose strukturor- selektiv (ndërkristalor). 8 Një shembull i KORROZIONIT SELEKTIV KOMPONENT është dezincifikimi i bronzit. Dezincifikimi i bronzit3 konsiston në faktin se zinku kalon në elektrolit (zakonisht në një vlerë pH neutrale ose pak acid) më intensivisht se bakri. Në sipërfaqen e bronzit formohet një shtresë e lirshme bakri, e cila kontribuon në rritjen e korrozionit elektrokimik (shfaqja e një qelize mikrogalvanike). Një shembull i korrozionit STRUKTURO-SELEKTIV është korrozioni i gize gri. Korrozioni strukturor selektiv i gizës gri përbëhet nga shkatërrimi mbizotërues i përbërësit të ferritit, duke rezultuar në formimin e një skeleti grafiti të mbushur me produkte korrozioni (Fig. 1−3). Në të njëjtën kohë, forca mekanike e gize zvogëlohet ndjeshëm. KORROZIONI INTERKRISTALITET karakterizohet nga shkatërrimi selektiv i metalit përgjatë kufijve të kokrrizave (Fig. 1-7). Si rezultat, me një ndryshim të vogël në masë dhe pamje, mund të ndodhë një humbje shumë e konsiderueshme e vetive të forcës së metalit (Fig. 2). 40 Fig. 2. Humbja e vetive të forcës së metalit gjatë llojeve të ndryshme të shkatërrimit nga korrozioni 1 – korrozioni uniform; 2 – korrozioni ndërgranular 20 Humbja e w, % 60 2 0 1 2 4 Humbje peshe, kg/cm2 Në raste ekstreme, me shkatërrimin e plotë të kufijve të kokrrizave si rezultat i korrozionit ndërgranular, aliazhi mund të shkërmoqet në pluhur. PRARITJA E KORROZIONIT është një rast i korrozionit lokal në të cilin shkatërrimi ndodh në drejtimin pingul me sforcimin më të madh tërheqës (Fig. 1 - 8). 3 Tunxh – aliazh bakër-zink. 9 Është tipike që në këtë rast një çarje korrozioni mund të përhapet përgjatë kufijve të kokrrizave ose të presë trupin kristal. Shkatërrimi nga lodhja nga korrozioni (me ekspozim të njëkohshëm ndaj një mjedisi gërryes dhe ngarkesave alternative ose ciklike) zhvillohet në mënyrë të ngjashme si gjatë plasaritjes nga korrozioni. KORROZIONI NËNSIFORËS, duke filluar nga sipërfaqja, përhapet kryesisht nën sipërfaqe, gjë që shpesh çon në shtrembërim dhe ënjtje të metalit (Fig. 1− 9), për shembull, në formimin e flluskave në fletë metalike me cilësi të ulët gjatë procesit të gravurës. ose korrozioni gjatë funksionimit të enëve dhe aparateve. 1.4. Treguesit e korrozionit Për të përcaktuar shkallën e korrozionit përdoren treguesit e korrozionit: MASSOMETRIK, VOLUMETRIK, THELLËSIM, MEKANIKE, RRYMË. 2 Indeksi i korrozionit MASSOMETRIK (Km, g/m ∙h) është ndryshimi i masës metalike si rezultat i korrozionit për njësi sipërfaqe (s) të kampionit dhe për njësi të kohës. Ndryshimi në masën e një kampioni zakonisht përcaktohet si diferenca midis masës së mostrës para testimit (mо) dhe masës së saj pas testimit (m1) me kalimin e kohës (τ) me heqjen e produkteve të korrozionit (humbja e masës metalike) . K m = (m0 − m1)/s ∙ τ . (4) 3 2 Indeksi VOLUMETRIK i korrozionit (Kν, cm /m h) është vëllimi (Vo) i gazit të përthithur ose të çliruar gjatë procesit të korrozionit, për njësi sipërfaqe metalike dhe për njësi të kohës K m = V0 /s ∙ τ. (5) Indeksi i korrozionit DEPTH (P, mm/vit) merr parasysh uljen e trashësisë së metalit për shkak të korrozionit, të shprehur në njësi lineare dhe për njësi të kohës. Ky tregues është i dobishëm kur krahasohet korrozioni i metaleve me dendësi të ndryshme. Kalimi nga treguesi masometrik në thellësi mund të bëhet, në rastin e korrozionit uniform, duke përdorur formulën P = K m ∙ 8,76 /γ, (6) ku γ është dendësia e metalit. Treguesi MEKANIK i korrozionit përcaktohet nga ndryshimi i njërit prej treguesve kryesorë të vetive mekanike të një metali gjatë një kohe të caktuar të procesit të korrozionit dhe shprehet në njësi relative ose në përqindje. Nëse forca në tërheqje përdoret si tregues mekanik, atëherë ndryshimi në treguesin e forcës (Kσ) përcaktohet me formulën Kσ = · Δσb/σb, (7) ku σb është forca në tërheqje përpara korrozionit, Δσb është ndryshimi i qëndrueshmërisë në tërheqje të metalit pas korrozionit gjatë kohës τ. Treguesi aktual i korrozionit lejon, duke përdorur formulën e Faradeit, të llogarisë sasinë e metalit të gërryer bazuar në madhësinë e rrymës së korrozionit Km = IA /nF, (8) ku Km është shkalla e korrozionit bazuar në ndryshimin në masë metalike; I – forca aktuale; F – konstante Faraday; n – valencë metalike; A është masa atomike e metalit. Gjatë vlerësimit cilësor dhe sasior të rezistencës ndaj korrozionit të metaleve, rekomandohet të përdoret një shkallë me dhjetë pikë4. Duhet të theksohet se, megjithëse kjo shkallë për vlerësimin e rezistencës ndaj korrozionit të metaleve është bërë më e përhapur, veçanërisht në industrinë kimike, ajo nuk është universale, pasi për pajisjet për qëllime të ndryshme, shkalla e lejueshme e korrozionit mund të ndryshojë me një renditje të madhësisë ose më shumë. Tabela 1 Dhjetë -Shkalla ballike e rezistencës ndaj korrozionit të metaleve dhe lidhjeve. në 0,1 mbi 0,1 në 0,5 mbi 0,5 në 1,0 mbi 1,0 në 5,0 mbi 5,0 në 10,0 mbi 10,0 REZULTATI 1 2 3 4 5 6 7 8 9 10 Sistemi i unifikuar i mbrojtjes kundër korrozionit dhe plakjes. Strukturat nëntokësore. Kërkesat e përgjithshme për mbrojtjen nga korrozioni. GOST 9.602−2005, http://www.gosthelp.ru/gost/gost206.html 4 11 Kapitulli 2. KORROZIONI KIMIK I METALEVE Korrozioni kimik i metaleve është një proces spontan i shkatërrimit të tyre për shkak të ndërveprimit kimik me mjedisin e jashtëm. Karakteristikat dalluese të procesit të korrozionit kimik janë: – zbatimi i proceseve të oksidimit dhe reduktimit në një fazë; - mungesa e rrymës elektrike; – formimi i produkteve të korrozionit direkt në sipërfaqen ku ndodh shkatërrimi i tij. KORROZIONI KIMIK është një reaksion heterogjen i ndërveprimit ndërmjet fazës së lëngshme (l) ose gazit (g) me fazën e ngurtë (s) - metali dhe ndahet në 2 lloje (korrozioni me gaz, korrozioni në jo-elektrolite të lëngëta). 2.1. Korrozioni i gazit Në praktikë, rëndësia më e madhe është KOROZIONI I GAZIT. Më shpesh, ajo manifestohet si korrozioni i materialeve metalike në temperatura të larta (dhjetëra herë më të larta se pika e vlimit të ujit) në një atmosferë të gazrave që përmbajnë oksigjen. Për këtë arsye quhet KORROZION NË TEMPERATURË TË LARTË ose OXIDIM NË TEMPERATURË TË LARTË. Në sipërfaqen e produkteve të bëra nga metale dhe lidhje, si rezultat i ndërveprimit të tyre me një mjedis të tillë gërryes, formohet një film produktesh që përfaqësojnë okside të ndryshme metalike. Në të njëjtën kohë, në varësi të kushteve të funksionimit, mund të formohen komponime të tjera, për shembull, metale squfuri5. Për shembull, në industrinë e rafinimit të naftës, korrozioni i gazit prek, për shembull, tubat e furrave, muret e reaktorit, kolonat e reagimit dhe pajisjet e tjera të ekspozuara ndaj sulfurit të hidrogjenit, monoksidit të karbonit, azotit, oksigjenit dhe gazrave të tjerë në temperatura të larta. Termi "korrozioni i gazit" i referohet shkatërrimit të metaleve të shkaktuar nga veprimi i avujve dhe gazeve në temperatura të larta, megjithëse mund të aplikohet edhe në temperatura më të ulëta në 5 METALE TË SQUFURIT, njësoj si sulfuri. 12 sigurohuni që një film lëngu që përcjell rrymë elektrike të mos kondensohet në metal. Në sipërfaqen e produkteve të bëra nga metale dhe lidhje, si rezultat i ndërveprimit të tyre me një mjedis gërryes, formohet një film produktesh që përfaqësojnë komponime të ndryshme metalike. Në përgjithësi, procesi i korrozionit kimik mund të përshkruhet, për shembull, nga reaksioni i oksidimit të metaleve me gaz të nxehtë (T >>373K) (G2): 2Ме + nГ2 → 2Men+Гn. Në një mjedis oksigjeni, një proces i tillë mund të përshkruhet nga ekuacioni i mëposhtëm Me(T) + ½ O2 ↔ MeO(T). Ky reagim është ekuilibër në temperatura të larta. Procesi i kundërt quhet SHKARTËSIMI I OXIDIT. Oksigjeni i pranishëm në një mjedis të gaztë karakterizohet nga presioni i tij i pjesshëm (pO2). Reaksioni kimik i oksidimit të metaleve do të jetë në ekuilibër me reaksionin e shpërbërjes së oksidit nëse presioni i pjesshëm i oksigjenit (pO2) dhe ELASTICITETI I SHPËRNDARJES SË OXIDIT (pMeO)7 bëhen të barabarta (pMeO = pO2) 1. Kur pO2 > pMeO, reaksioni vazhdon. kryesisht në drejtim të formimit të oksidit. Kur pMeO > pO2, reaksioni vazhdon kryesisht në drejtim të kundërt. Oksidimi i metaleve është një proces heterogjen me shumë faza, për shkak të të cilit së pari formohet një shtresë oksidi mono- dhe më pas një polimolekulare, i ashtuquajturi film oksid. Rritja e trashësisë së tij mund të ndodhë ose në të dy drejtimet njëkohësisht (metal dhe i gaztë), ose kryesisht në një (Fig. 3). PRESIONI I PJESSHËM (nga latinishtja e vonë partialis - i pjesshëm), presioni që do të kishte një gaz i përfshirë në një përzierje gazi nëse i vetëm do të zinte një vëllim të barabartë me vëllimin e përzierjes në të njëjtën temperaturë. 7 Presioni i pjesshëm i ekuilibrit të oksigjenit për reaksionet e formimit ose SHPËRNDARJES TERMIKE të OKSIDEVE quhet ELASTICITETI I SHKARTËSIMIT TË OKSIDEVE. Vlera e tij lidhet me konstantën e ekuilibrit të reaksionit dhe, për rrjedhojë, është një funksion i temperaturës. 6 13 a b c Fig. Fig. 3. Skema e mekanizmit jono-elektronik të formimit dhe rritjes së një filmi oksid në sipërfaqen e një pjese metalike Nëse rritja ndodh në drejtim të një mjedisi të gaztë, atëherë vërehet një rritje e konsiderueshme në madhësinë e pjesës , për shembull, gjatë oksidimit, i cili duhet të merret parasysh në inxhinierinë mekanike. Drejtimi i rritjes përcaktohet nga raporti ndërmjet shkallëve të kundër-difuzionit të joneve metalike Мen+ dhe oksigjenit O2− brenda filmit oksid. Nëse shkalla e difuzionit të joneve metalike (rdif.Me) dhe oksigjenit (rdif.O2) ndryshojnë shumë, atëherë rritja e filmit të oksidit ndodh kryesisht në një drejtim, për shembull, me rdiff.O2< rдиф.Mе − в направлении газовой среды и наоборот (рис. 3, в, б). В большинстве случаев, скорости диффузии ионов металла и кислорода соизмеримы, но для ионов кислорода скорость несколько ниже из−за того, что они больше по размерам, поэтому зона роста, находящаяся в толще самой оксидной пленки, располагается ближе к ее внешней поверхности (рис. 3, а). По мере утолщения оксидной пленки процессы встречной диффузии ионов и электронов затрудняются. С повышением температуры упругость диссоциации оксидов возрастает у всех металлов. Поэтому, начиная от какой−то температуры, когда упругость диссоциации оксида металла становится больше парциального давления кислорода в воздухе (при атмосферном давлении рО2 = 0,21 ат), металл перестает окисляться и становится вполне малоактивным по отношению к кислороду. Так, например, серебро при температуре 300 ºК термодинамически еще не устойчиво, а при 400 ºК и при всех более высоких температурах упругость диссоциации Ag2O превышает парциаль14 ное давление кислорода в воздухе. При температуре 2000 ºК медь тоже становится неокисляемым металлом, однако для таких металлов, как Fе, Zn, Ni и др., даже при этих температурах упругость диссоциации окислов остается еще достаточно низкой и, следовательно, протекание реакции окисления вероятно. Необходимо учесть, что температурная зависимость для кинетики процесса окисления совершенно иная, чем для термодинамической вероятности окисления. Поэтому нет никакого противоречия в том, что термодинамическая вероятность окисления металла с повышением температуры падает, а реальная скорость окисления (при условии, что термодинамически этот процесс остается еще возможным) с повышением температуры возрастает. 2.2. Образование тонких пленок на металлах Ранее было отмечено, что в большинстве случаев образовавшиеся продукты газовой коррозии – оксиды металла остаются на поверхности металла в виде пленки. Эти пленки определяют кинетику процесса и в случае наличия защитных свойств могут привести к замедлению коррозии. Чтобы пленка оксида металла обладала защитными свойствами, она должна быть: − сплошной; − прочно сцепляться с основным металлом и обладать близким к нему коэффициентом теплового расширения; − быть химически инертной по отношению к данной агрессивной среде; − обладать твердостью и износостойкостью. Даже при обычной температуре на поверхности многих металлов и сплавов на воздухе образуются слои оксидов. Еще в 1836 г. М. Фарадей высказал предположение, что даже на блестящей поверхности металлов существует тончайшая, невидимая защитная (оксидная) пленка. Этим он объяснял пассивность железа в азотной кислоте. Если пленка пористая, рыхлая и характеризуется плохим сцеплением с основным металлом, то даже при условии инертности в данной агрессивной среде, она не будет обладать защитными свойствами. Пленки по толщине (δ) делятся на три группы: − тонкие (невидимые), δ < 4000 нм; − средние (дающие цвета побежалости), δ = от 4000 до 50000 нм; 15 − толстые (видимые), δ > 50000 nm. Trashësia e filmave të formuar gjatë bashkëveprimit të metalit me ajrin e thatë është e ndryshme dhe varet nga lloji i metalit, temperatura dhe faktorë të tjerë. Kështu, trashësia e filmave në bakër dhe hekur në temperaturën e dhomës është 100-300 nm, dhe në alumin 1000-1500 nm. Le të japim disa shembuj që vërtetojnë praninë e shtresave të hollë në metale. 1. Kur sipërfaqja e pastër me shkëlqim e një pllake hekuri nxehet në njërin skaj, do të shfaqen ngjyrat e ndërhyrjes - "njollat e ngjyrave". Kjo tregon shfaqjen e oksideve me trashësi të ndryshme. Ngjyrat shkojnë në rendin e mëposhtëm - nga fundi i ftohtë i pjatës në atë të nxehtë, përkatësisht: e verdhë, portokalli, e kuqe, vjollcë, vjollcë, blu. Gjatë testimit të vetive mbrojtëse të filmit të formuar duke përdorur pika të një solucioni të nitratit të bakrit, në vendet ku filmi ka veti të dobëta mbrojtëse, ai reagon me hekurin: Cu(NO3)2 + Fe → Fe(NO3)2 + Cu. Si rezultat, bakri do të shfaqet në sipërfaqe nën pika. Deri në kohën pas së cilës ndodh reagimi, mund të gjykohen vetitë mbrojtëse të filmave në zona të ndryshme. Në këtë eksperiment, vetitë më të mira mbrojtëse gjenden në zonën që ndodhet larg vendit të ngrohjes (prapa zonës së verdhë), ku sipërfaqja mbetet e pandryshuar. Është e qartë se në këtë zonë ka një film shumë të hollë, por më të dendur në krahasim me zonat e nxehta. 2. Nëse thyeni një gjilpërë (ose brisk) nën merkur, në pikat e thyera do të formohet amalgamë. Kur mostrat e thyera vendosen në ajër në merkur, bashkimi nuk vërehet. Kjo shpjegohet me formimin e një filmi shumë të hollë oksidi, i cili parandalon shkrirjen. 3. Kur hekuri vendoset në një tretësirë të hidroksidit të kaliumit me jod të tepërt, në sipërfaqen e hekurit në tretësirë bëhet një gërvishtje e thellë. Pas ca kohësh, hekuri në vendin e gërvishtjes fillon të shpërndahet dhe thekon mbeten në fund të xhamit, të cilat pas ekzaminimit të mëtejshëm rezultojnë të jenë oksid hekuri (III). Procesi i formimit të filmit në metal ndodh në faza. 16 Faza e parë e procesit të bashkëveprimit të një metali me një mjedis gërryes është kimisorbimi i agjentit oksidues (O2, CO2, H2O, SO2) në sipërfaqen e metalit, për shembull: Me(sol) + (O2) = Me( sol) + 2Oads. Në prani të afinitetit kimik midis metalit dhe agjentit oksidues, realizohet faza e dytë e bashkëveprimit të metalit me mjedisin gërryes - kalimi i filmit të kimisorbuar në atë oksid. Ky proces mund të përshkruhet në mënyrë konvencionale nga një reagim i formës: xMe(s) + yOads = MexOy. Filmi oksid i formuar në sipërfaqen e metalit mund të ngadalësojë procesin e korrozionit për shkak të frenimit të furnizimit të agjentit oksidues në sipërfaqen e metalit oksidues. Në këtë rast, filmi KA VETITË MBROJTËSE. 2.2.1. Kushti për vazhdimësinë e filmave në metale Vetëm FILMAT E VAZHDUESHËM mund të kenë veti mbrojtëse. Mundësia e formimit të një filmi të tillë përcaktohet nga KUSHTI I VAZHDIMËSISË Pilling–Bedworth. Vëllimi molar i përbërjes që shfaqet në sipërfaqen e metalit (Vok) duhet të jetë më i madh se vëllimi i metalit (VMe) i konsumuar për të formuar një mol të përbërjes: Vok/VMe >1. Nëse Vok/VMe< 1, то пленка не может быть сплошной. К металлам, не удовлетворяющим условию сплошности при окислении их кислородом, относятся щелочные и щелочно−земельные металлы. Сплошность пленок – необходимый, но недостаточный фактор, определяющий ее защитные свойства. Как показал Францевич И. Н., у пленок с Vok/VMe >>1 nuk mund të ketë veti të larta mbrojtëse, për shkak të shfaqjes së sforcimeve të mëdha të brendshme në to (për shembull, MoO3 ose WO3). Kufiri i sipërm i raportit të volumit merret si 2.5. Atëherë gjendja e vazhdimësisë së rafinuar duket si: 1< Vok/VMe < 2,5. 2.2.2. Законы роста оксидных пленок на металлах Процесс роста ПОРИСТОЙ ПЛЕНКИ не осложняется процессом подвода окислителя к поверхности металла, а контролируется скоростью химической реакции образования оксида. Скорость коррозии в этом случае выражается уравнением: 17 V = dx/d = kC, где x − толщина пленки; – время; k − константа скорости химической реакции образования оксида; C − концентрация окислителя на поверхности метала. Рост пористой пленки, контролируемый скоростью химической реакции окисления металла, протекает во времени ПО ЛИНЕЙНОМУ ЗАКОНУ. Линейный закон роста имеет место: − при высокотемпературном окислении на воздухе и в среде кислорода при Vok/VMe<1; − в случае образовании летучих оксидов. Сплошные пленки затрудняют проникновение реагентов друг к другу и их рост сопровождается самоторможением процесса. Результирующая скорость этого сложного процесса определяется скоростью самой медленной стадии, т.е. возможны различные варианты контроля течения процесса. 1−й вариант. Скорость коррозии контролируется стадией массопереноса (диффузионный контроль процесса). В этом случае концентрация окислителя на границе раздела Ме – оксидная пленка равна нулю, так как скорость химической реакции весьма велика. Уравнение скорости течения коррозии определяется ПАРАБОЛИЧЕСКИМ ЗАКОНОМ роста оксидной пленки. Параболический закон роста реализуется, в частности, при окислении железа на воздухе при различных температурах. 2−ой вариант. В предыдущем случае С = 0, т.е. накопление окислителя на внутренней поверхности оксидной пленки не происходит. Если учесть возможность кинетического торможения процесса роста оксидной пленки наряду с торможением на стадии массопереноса (диффузионно−кинетический контроль процесса), в этом случае приходим к СЛОЖНО − ПАРАБОЛИЧЕСКОМУ ЗАКОНУ роста оксидной пленки. Рост оксидных пленок по сложно−параболическому закону выполняется: а) при окислении железа в парах воды или в атмосфере СО2; б) при окислении Сo, Сu в атмосфере чистого кислорода; в) при частичном разрушении оксидной пленки. Рост оксидных пленок при диффузионно−кинетическом контроле может быть выражен ТАКЖЕ СТЕПЕННЫМ ЗАКОНОМ. 18 3−й вариант. Часто рост пленки протекает медленнее, чем это следует из параболического закона роста. Это наблюдается при возникновении оксидных пленок при низких температурах (на Сu в О2 при t < 100 оС; на Аl, Тi, Ni, Zn в О2 при t = 300400 оС.) Затухание объясняется либо уплотнением пленок, либо появлением дефектов (пузырей, расслоений). В этих случаях рост толщины пленки протекает в соответствии с логарифмическим законом роста: x = ln(K). 2.3. Термодинамика процесса химической коррозии В качестве примера рассмотрим процесс окисления металла в атмосфере кислорода. Процесс окисления металла может быть представлен реакцией вида: 2Ме + nО2 → Me2n+Оn. Движущая сила процесса коррозии (ДСП) может быть определена с помощью термодинамики. Торможение процесса (ТП) не может быть определено термодинамически, однако с помощью термодинамики можно оценить условия, уменьшающие или исключающие протекание процесса (применение защитных сред, катодная защита, обескислороживание и др.). Независимо от механизма коррозии возможность ее протекания определяется знаком изменения термодинамического потенциала (см. раздел 1.1). Любое изменение энергии системы характеризует переход ее в новое состояние и является мерой стабильности системы: ΔG = GII − GI , где ΔG – энергия, израсходованная на изменение состояния системы, например, на процесс коррозии; GI − энергия системы в исходном состоянии, например, металла в конкретных условиях; G II − энергия системы в новом состоянии, например, прокорродировавшего металла. Рассчитать ΔG можно, вспомнив начало химии8. Величину изобарно-изотермического потенциала можно рассчитать по разности между суммами Δ G продуктов реакции и исходных веществ: GХР = ΔG ni – ΔG mi. (прод. р-ции) (исх. в-в). ΔG - является функцией температуры. Стандартные значения для большинства веществ сведены в справочные таблицы термодинамических величин. Изменение энергии Гиббса в зависимости от температуры учитывается уравнением Гиббса. ΔG Т = ΔН 298 – 298 Δ S 298. ΔG простого вещества = 0. Определить энергию Гиббса для любой химической реакции можно по уравнению ΔG = ΔG + RТ ln К, рассчитав предварительно величину константы равновесия, например, по равновесным концентрациям веществ. При равновесии ΔG=0, поэтому ΔG+RТlnК=0, следова8 19 2.4. Кинетика процесса химической коррозии Хотя термодинамика и дает ответ на вопрос о том, насколько изучаемая система отдалена от состояния равновесия, но ответ на весьма важный практический вопрос о скорости протекания коррозионного разрушения в рамках термодинамики получен быть не может. Рассмотрением этого вопроса и занимается кинетика коррозионных процессов. 2.4.1. Скорость процесса коррозии. Показатели химической коррозии СКОРОСТЬ КОРРОЗИОННОГО процесса может быть представлена в общем виде с помощью уравнения: СКОРОСТЬ КОРРОЗИИ (СК) = ДСП / ТП, где ДСП − движущая сила процесса; ТП − торможение процесса. Скорость химической коррозии определяют количественно по наблюдениям во времени за изменением какой−либо подходящей для этих целей величины, изменяющейся в процессе коррозии. Тогда истинная скорость коррозии металлического материала определяется из выражения: V = dy/d, где y − изменяющаяся в процессе коррозии характеристика свойств материала; − время коррозии. Для количественного выражения скорости коррозионных разрушений на практике пользуются показателями коррозии, являющимися, по сути, отражением СРЕДНЕЙ СКОРОСТИ КОРРОЗИОННОГО разрушения материала: Vср=∆y/∆. Показатели коррозии. 1. ГЛУБИННЫЙ ПОКАЗАТЕЛЬ коррозии: Kn=∆П/∆, где ∆П − глубина (средняя или максимальная) коррозионного разрушения; ∆ − промежуток времени коррозии. 2. ПОКАЗАТЕЛЬ ИЗМЕНЕНИЯ ТОЛЩИНЫ образующейся на металле пленки продуктов коррозии: Kn= ∆h/∆, где ∆h − изменение толщины образующейся на металле пленки продуктов коррозии. тельно, ΔG = – RТlnК. Данное уравнение позволяет также определить значение константы равновесия химической реакции по величине изобарно-изотермического потенциала. Для определения направления протекания многих химических реакций достаточно оценить знак Δ G. 1). При ΔН 0 , Δ S 0 реакция самопроизвольна (Δ G 0) при Т. 2). При ΔН 0, Δ S 0 реакция обратная (Δ G 0) при любой Т. 3). При ΔН 0, Δ S 0, реакция самопроизвольна (Δ G 0) при любой Т. 4). При ΔН 0, ΔS 0 реакция самопроизвольна (Δ G 0) при Т. 20 3. ПОКАЗАТЕЛЬ ИЗМЕНЕНИЯ МАССЫ Km=∆m/S∆, где ∆m − изменение массы корродирующего металла; S – площадь поверхности коррозии. 4.ОБЪЕМНЫЙ ПОКАЗАТЕЛЬ КОРРОЗИИ Kv=∆V/S∆, где ∆V − объем газа, выделившегося или поглощенного в процессе коррозии и приведенный к нормальным условиям. 5. МЕХАНИЧЕСКИЙ ПОКАЗАТЕЛЬ КОРРОЗИИ Кs=(∆s/∆)100 %, где ∆s − относительное изменение характеристики механического свойства. 6. ПОКАЗАТЕЛЬ ИЗМЕНЕНИЯ ЭЛЕКТРИЧЕСКОГО СОПРОТИВЛЕНИЯ KR=(∆R/∆)100 %, где ∆R − относительное изменение электросопротивления образца. 2.4.2. Влияние внешних факторов на химическую коррозию металлов Скорость и характер процесса химической коррозии металлов зависит от ряда факторов. ВНЕШНИМИ ФАКТОРАМИ называют те, которые связаны с составом коррозионной среды и условиями коррозии (температура, давление, скорость перемещения коррозионной среды и т.д.). Температура − мощный внешний фактор коррозии. Характер влияния температуры на скорость окисления металла определяется зависимостью константы скорости реакции окисления (k) и диффузии (D) от температуры. И k = f(T) и D = f(T) описываются одним и тем же уравнением (уравнение Аррениуса): k = k0 exp (−E/RT), (9) где k0 − константа; Е − энергия активации химической реакции. D = D0 exp (−E1/RT), (10) 1 где D0 – константа; Е − энергия активации диффузии. Таким образом, вне зависимости от вида контролирующей стадии процесса окисления с повышением температуры скорость окисления резко возрастает. Колебания температуры, особенно переменный нагрев и охлаждение, увеличивают скорость окисления металла, так как в защитной пленке образуются трещины. Состав газовой фазы. Влияние состава газовой фазы на скорость коррозии металла велико, специфично и изменяется с изменением температуры. В частности, на скорость окисления железа и 21 стали особенно сильно влияют кислород, соединения серы, водяные пары. Приведенные ниже данные свидетельствуют о зависимости увеличения относительной скорости коррозии стали с 0,17 % углерода от состава газовой фазы при 900 оС: Состав газовой фазы Кислород Чистый воздух Чистый воздух + 2 % SO2 Чистый воздух + 5 % H2O Чистый воздух + 5 % SO2+ 5 % H2O % 200 100 118 134 276 Значительное влияние на коррозию сталей и сплавов оказывают продукты горения топлива, содержащие ванадий (например, V2O5). Это соединение находится в золе от сжигания дешевого топлива − мазута, нефтепродуктов. Зола, налипая на металл, увеличивает скорость его коррозии в десятки раз. Причина этому – «ванадиевая коррозия», обусловленная легкоплавкостью V2O5, и его способностью ОФЛЮСОВЫВАТЬ (переводить в жидкое состояние) химические соединения золы и окалины. Пятиокись ванадия участвует в процессе окисления железа: 4Fe + 3V2O5 = 2Fe2O3 + 3V2O3, V2O3 + O2 = V2O5, V2O5 + Fe2O3 = 2FeVO4. Повышение содержания СО в газовой фазе понижет скорость коррозии углеродистых и низколегированных сталей, но при больших количествах СО в газовой фазе может произойти науглероживание поверхности стали. При этом возможны следующие химические реакции: 2CO + O2 = 2CO2, 2CO = Cсаж + CO2. Давление окислителя. Если бы поверхность металла не была покрыта оксидной пленкой, то скорость окисления была бы пропорциональна парциальному давлению окислителя (для двухатомных газов – PГ2). Так как поверхность металла покрыта слоем оксида, то зависимость скорости окисления от величины парциального давления окисляющего газа определяется строением защитной пленки. В то же время при окислении железа при 700 − 950о С скорость окисления не зависит от парциального давления Po2. 22 Скорость движения газовой фазы. Окисление как гетерогенный процесс определяется скоростью подвода и отвода реагентов в зону реакции. Поэтому, чем больше скорость движения потока газа, тем больше и скорость окисления металла. Режим нагрева металла. Влияние режима нагрева металла может быть рассмотрено в зависимости от влияния колебаний температуры. Иначе говоря, переменные нагрев и охлаждение увеличивают скорость окисления ввиду нарушения сохранности защитной пленки. 2.4.3. Влияние внутренних факторов на химическую коррозию металлов ВНУТРЕННИМИ называют факторы, связанные с составом, структурой, внутренними напряжениями в металле, качеством обработки поверхности металла и др. Состав сплава. Применительно к наиболее важным конструкционным сплавам на основе железа для случая газовой коррозии можно отметить следующее. 1. При высоких температурах (более 800оС) с увеличением содержания углерода в стали скорость ее окисления и обезуглероживания уменьшается вследствие более активного образования СО, что снижает окислительный потенциал газовой фазы. 2. Сера фосфор, никель, марганец практически не влияют на скорость окисления железа. 3.Титан, медь, кобальт, бериллий заметно снижают скорость окисления железа. 4. Хром, алюминий, кремний сильно замедляют окисление железа. 5. Ванадий, вольфрам и молибден могут значительно ускорить окисление стали, которое иногда носит катастрофический характер. Структура сплава. Анализ экспериментальных данных свидетельствует о том, что чем меньше в сплаве структурных составляющих, тем выше его жаростойкость. Применительно к сплавам железо – углерод наиболее устойчивой является АУСТЕНИТНАЯ СТРУКТУРА9. Аустенит (γ-фаза) − высокотемпературная гранецентрированная модификация железа и его сплавов. В углеродистых сталях аустенит − это твѐрдый раствор внедрения, в котором атомы углерода входят внутрь элементарной ячейки γ-железа во время конечной термообработки. В сталях, содержащих другие металлы (кроме железа, легированные стали), атомы металлов замещают атомы железа в кристаллической решетке, и возникает твердый раствор замещения. Фаза названа в честь сэра Уильяма Чандлера Робертс-Остина (англ. William Chandler Roberts-Austen, 1843 − 1902). 9 23 Всего различают пять больших групп нержавеющих сталей, определяемых их микроструктурой. Наиболее распространенными являются три: АУСТЕТНИТНЫЕ (AUSTENITIC) − не магнитная сталь с основными составляющими 15−20 % хрома и 5−15 % никеля, который увеличивает сопротивление коррозии. Она хорошо подвергается тепловой обработке и сварке. Обозначается буквой A в начале наименования марки. Именно аустенитная группа сталей наиболее широко используется в промышленности и в производстве элементов крепежа. МАРТЕНСИТНЫЕ (MARTENSITIC) − значительно более твердые, чем аустетнитные стали и могут быть магнитными. Они упрочняются, закалкой и отпуском подобно простым углеродистым сталям и находят применение главным образом в изготовлении столовых приборов, режущих инструментов и общем машиностроении. Больше подвержены коррозии. Обозначаются буквой С. ФЕРРИТНЫЕ (FERRITIC) стали значительно более мягкие чем мартенситные по причине малого содержания углерода. Они также обладают магнитными свойствами. Обозначаются буквой F. Стали с двухфазной АУСТЕНИТНО−ФЕРРИТНОЙ СТРУКТУРОЙ менее устойчивы против окисления. Их меньшая жаростойкость связывается с большей неоднородностью образующейся защитной пленки, что приводит к ее разрушению при росте (неоднородность возникающих внутренних напряжений). Так хромоникелевые стали с однофазной аустенитной структурой более устойчивы против окисления, чем двухфазные: Х12Н12М2Т, Х12Н9Т ведут себя лучше, чем ОХ21Н5МД2Т или 1Х21Н5Т 2]. На жаростойкость10 чугунов кроме их состава влияет и структура. Существенное влияние оказывает форма графитных включений. Так, при шаровидной форме графита стойкость против окисления выше, чем при пластинчатой. Установлено, что предварительная холодная пластичная деформация несколько ускоряет окисление металла вследствие повышения запаса его энергии. Чем тщательнее обработана поверхность металла, тем меньше скорость его окислеЖаростойкость − окалиностойкость, способность металлических материалов противостоять химическому разрушению поверхности под воздействием воздушной или иных газообразных сред при высоких температурах. 10 24 ния, что обусловлено лучшей сохранностью защитных пленок на гладкой поверхности. 2.5. Некоторые случаи газовой коррозии металлов в технологических средах В химической промышленности многие технологические процессы или их определенные стадии протекают в газовой среде в условиях повышенных температур и давлений (рис.4). При температурах от 100 до 200 − 300 °С многие газы не опасны. Химическая активность газов и скорость газовой коррозии металлов сильно возрастают при температурах выше 200 − 300 °С. Так, Cl2 начинает действовать на железные сплавы при Рис. 4. Некоторые случаи газовой температуре > 200 °C, HCl - > korrozioni i metaleve në teknologji - 300 °C, SO2, NO2, avujt e squfurit. > 500 °C. Këto veçori të sjelljes së mjediseve teknologjike të gazit dhe përdorimi i tyre i gjerë në industri kërkojnë një shqyrtim më të detajuar të sjelljes së metaleve në kushte reale. 2.5.1. Korrozioni i hekurit, gizës dhe çeliqeve në një atmosferë avujsh O2, CO2, H2O Ndikimi i oksigjenit të ajrit në temperaturat e larta në lidhjet hekur-karbon çon në oksidimin e hekurit me formimin e shkallës, dekarburizimin e çelikut dhe rritjen. prej gize. Si rezultat i oksidimit të hekurit në temperatura të larta, formohet një shtresë e produkteve të korrozionit të quajtur SCALE. Shkalla ka një strukturë komplekse dhe përfshin disa okside: Fe3O4 – MAGNETI ka një rrjetë kristalore komplekse spineli; Fe2O3 – HEMATITI ka një rrjetë romboedrale; FeO – VUSTIT ka një strukturë të dëmtuar të rrjetës kristalore. Wustiti formohet në temperatura mbi 575 oC dhe dekompozohet me ftohje të ngadaltë: 4FeO Fe3O4 + Fe. 25 Nën 575 °C nuk ka wustit në shkallë dhe një shtresë Fe3O4 është drejtpërdrejt ngjitur me sipërfaqen e çelikut. Shpejtësia e oksidimit të çelikut (shih seksionin 2.4.1) rritet me rritjen e temperaturës sipas një ligji afër eksponencialit (Fig. 5), dhe në koordinatat logk − 1/T shprehet me një vijë të thyer, çdo thyerje e së cilës korrespondon me disa transformime. Kështu, në një temperaturë prej 575 ºC, në shkallë shfaqet një nënshtresë e wustit FeO, e cila nuk ndërhyn në difuzionin e oksigjenit, si rezultat i së cilës rritet energjia e aktivizimit të procesit dhe rritet shkalla e oksidimit të metaleve. Thyerja në kurbë në një temperaturë prej ~ 900 ºС korrespondon me transformimin alotropik të çelikut. Natyra e ndryshimit të varësisë nga temperatura e shkallës së oksidimit të çelikut tregon se struktura austenitike është më rezistente ndaj nxehtësisë, në të cilën vërehet një rritje më e ngadaltë e shkallës së oksidimit me rritjen e temperaturës. Së bashku me oksidimin në çelik dhe gize, ndodh procesi i dekarbonizimit - shterimi i shtresës sipërfaqësore në karbon për shkak të ndërveprimit të karbitit të hekurit që përmbahet në to me oksigjen dhe reagjentë që përmbajnë oksigjen Fe3C+O2→3Fe+CO2 Fe3C+CO2. →3Fe+2CO Fe3C+H2O→3Fe+CO + H2. 26 Përbërja e mjedisit të gazit ka një ndikim të fortë në shkallën e oksidimit të çelikut dhe gize. Në korrozionin e gazit, agjentët agresivë mund të jenë, për shembull, klori, komponimet e squfurit, oksigjeni, ajri, përbërjet e jodit, etj. Agresiviteti i tyre ndaj metaleve të ndryshme nuk është i njëjtë, prandaj, shkalla e korrozionit ndryshon. Kështu, shpejtësia e oksidimit të Fe, Co, Ni në një temperaturë prej 900 °C rritet në rendin H2O → CO2 → O2 → SO2. Në këtë rast, metalet, në varësi të shkallës së korrozionit në atmosferën e këtyre reagentëve, renditen në një seri ngjitëse: Ni → Co → Fe. Nëse supozojmë se shkalla e korrozionit në ajër në 900 °C është 100%, atëherë shtimi i 2% SO2 e rrit këtë shkallë në 118%, 5% H2O në 134%, 2% SO2 + 5% H2O në 276%. 2.5.2. Korrozioni nën ndikimin e produkteve të djegies së karburantit Produktet e djegies së karburantit (thëngjilli, nafta, etj.) në shumicën e rasteve përmbajnë sasi të konsiderueshme të përbërjeve të squfurit (SO2, H2S) dhe vanadiumit në formën e V2O5, i cili është një agjent i fortë oksidues. Oksidi i vanadiumit me shkrirje të ulët ndërvepron me shtresën mbrojtëse të shkallës në sipërfaqen e metalit, duke e shkatërruar atë dhe duke formuar vanadate, të cilat krijojnë eutektikë11 me pikë shkrirjeje të ulët Fe2O3 + V2O5 → 2FeVO4 me oksid vanadiumi V2O5. Kështu, vanadati i hekurit (FeVO4) ka pikë shkrirjeje = 840 °C, dhe pika e shkrirjes së eutektikës së tij me V2O5 është 625 °C; Pika e shkrirjes së vanadatit të kromit CrVO4 është 810 °C, dhe eutektika e tij me V2O5 është 665 °C. Pastaj vanadati merr pjesë aktive në procesin e oksidimit të vetë metalit 6FeVO4+4Fe→ 5Fe2O3 + 3V2O3 4Fe+ 3V2O5→ 2Fe2O3 + 3V2O3 V2O3+O2→ V2O5, dhe vetë V2O5 praktikisht nuk konsumohet. Në masën më të madhe, korrozioni i vanadiumit ndikon në mbështjelljet e furrës, mbështetëset dhe ndarjet që funksionojnë në intervalin e temperaturës 620 – 700 °C. Eutektik (nga greqishtja éutektos - shkrihet lehtë), një sistem i lëngshëm (tretësirë ose shkrirë) që është në një presion të caktuar në ekuilibër me fazat e ngurta, numri i të cilave është i barabartë me numrin e përbërësve të sistemit. 11 27 Në të njëjtën kohë, shkalla e korrozionit edhe të çeliqeve krom dhe krom-nikel, të cilët nuk janë të prirur ndaj korrozionit të sulfurit të hidrogjenit në temperaturë të lartë, arrin 15 mm/vit. Nën ndikimin e komponimeve të squfurit, çeliqet hekur-karbon i nënshtrohen korrozionit intensiv ndërgranular për shkak të një numri më të madh të defekteve në rrjetat kristalore të sulfideve sesa oksidet. Kjo çon në intensifikimin e proceseve të difuzionit. Me një rritje të përmbajtjes së monoksidit të karbonit (II) në produktet e djegies, shkalla e korrozionit të gazit të karbonit dhe çeliqeve me aliazh të ulët zvogëlohet ndjeshëm. Megjithatë, përqendrimi i tij shumë i lartë çon në karburizimin e sipërfaqes: 3Fe+2CO→Fe3С+CO2. Lidhjet shumë të lidhura të tipit X40N50 janë rezistente ndaj korrozionit të vanadiumit. Futja e silikonit në çelik ka gjithashtu një efekt pozitiv aliazhimi i çelikut me molibden, tungsten dhe vanadium ka një efekt negativ në qëndrueshmërinë e çelikut, pasi këta elementë aliazh nxisin12 formimin e V2O5. Lidhjet me bazë Cu janë shumë të ndjeshme ndaj korrozionit të tillë. Për të reduktuar shkallën e korrozionit të vanadiumit, aktualisht përdoren këto: − kufizimi i temperaturës së funksionimit në 650 °C; - përdorimi i lidhjeve të tipit X40N50 për prodhimin e komponentëve që i nënshtrohen këtij lloji shkatërrimi; − fryrje në lëndë djegëse pluhur dolomiti që përmban okside magnezi dhe alumini, të cilat formojnë komponime refraktare me oksid vanadiumi; − duke kufizuar përmbajtjen totale të natriumit dhe vanadiumit në lëndë djegëse në jo më shumë se 2 10–4%. 2.5.3. Korrozioni në mjedisin e klorit dhe klorurit të hidrogjenit Sjellja e metaleve në mjedisin e klorit të gaztë dhe klorurit të hidrogjenit është thelbësisht e ndryshme nga veprimi i mjediseve të tjera agresive. Kjo për faktin se kripërat e klorurit që formohen në sipërfaqen e metalit kanë një pikë shkrirjeje të ulët dhe në disa raste kur temperatura rritet sublimohen13, për shembull Ti + 2Cl2 → TiCl4. Promoting - (sinonime) ngarje, promovim, drejtim. Sublimimi është kalimi i një lënde nga një gjendje e ngurtë në një gjendje të gaztë, duke anashkaluar fazën e lëngshme; njëjtë si sublimimi. 12 13 28 Shumica e këtyre reaksioneve janë ekzotermike. Shkalla e heqjes së nxehtësisë rezulton të jetë më e ulët se shkalla e vetë reagimit, si rezultat i së cilës metalet ndizen dhe "digjen" në një atmosferë klori. Kjo çon në një rritje të konsiderueshme lokale të temperaturës, dhe kloruret që rezultojnë shkrihen dhe dekompozohen. Më rezistentët ndaj klorit janë çeliqet e nikelit, plumbit dhe kromit. Temperatura e ndezjes së disa metaleve në një atmosferë me klor të thatë: o ndezje. Me Ti< 20 Pb 90−100 Fe 150 Cu 200 Ni > 500. Në klorurin e thatë të hidrogjenit në temperaturën e dhomës, një sërë metalesh dhe lidhjesh kanë rezistencë të kënaqshme. Me rritjen e temperaturës, rezistenca e materialeve metalike zvogëlohet gradualisht në një temperaturë specifike për secilin metal. Temperaturat maksimale të larta të lejueshme gjatë funksionimit afatgjatë të metaleve dhe lidhjeve në klorin e thatë dhe klorurin e hidrogjenit janë dhënë në tabelë. 2. Metalet më rezistente në klorin e thatë, me përjashtim të metaleve fisnike, janë nikeli dhe lidhjet e tij. Filmat sipërfaqësor të formuar në çelik nikel dhe krom-nikel kanë paqëndrueshmëri të ulët dhe veti të kënaqshme mbrojtëse. Tabela 2 Temperaturat e lejuara gjatë përdorimit të disa metaleve dhe lidhjeve në një atmosferë me klorur hidrogjeni dhe klori3] T, °C Materiali Cl2 1200 − − 550 − 100 150 300 − Platinum Gold Tungsten Nikel Inconel (80% Ni, 14 6% Fe) Bakër Karbon çeliku St.Z Inox 12Х18Н9Т Argjend 29 HCl 1200 870 600 510−600 480 100−120 260−350 450−500 230 2.5.4. Korrozioni me hidrogjen i çelikut Korrozioni hidrogjenor mund të shoqërojë shumë procese teknologjike që ndodhin në temperatura të larta nga 200°C dhe presione nga 300 MPa në mjedise që përmbajnë hidrogjen. Këto kushte korrespondojnë me procese të tilla si hidrogjenizimi i qymyrit dhe naftës, sinteza e amoniakut dhe metanolit, etj. Vërehen dy lloje të dëmtimit të metaleve nga hidrogjeni - HYDROGJEN BRITLESH dhe HYDROGJEN KORROZION. Shpesh këto dukuri mbivendosen me njëra-tjetrën. Nëse amoniaku është i pranishëm në gaz, atëherë mund të ndodhë edhe NITRIDIMI I METALIT. Kur një përzierje azot-hidrogjen bie në kontakt me një metal në kushte të temperaturave dhe presionit të ngritur, hidrogjeni molekular shpërndahet në sipërfaqen e metalit. Hidrogjeni atomik që rezulton shpërndahet në rrjetën metalike dhe shpërndahet në të. Ndërsa temperatura zvogëlohet për shkak të zvogëlimit të tretshmërisë, hidrogjeni tenton të shkojë në një gjendje të gaztë brenda metalit. Në këtë rast, streset e mëdha lindin në metal, duke çuar në brishtësinë e pakthyeshme. Korrozioni i hidrogjenit është rezultat i ndërveprimit kimik të hidrogjenit me përbërësin karabit të çelikut. Nga jashtë, shfaqja e korrozionit të hidrogjenit nënkupton një rënie të fortë të forcës së çelikut pa shkatërrim të dukshëm të sipërfaqes. Mekanizmi i korrozionit të hidrogjenit përfshin këto faza: - në temperatura të larta, hidrogjeni molekular shpërndahet në atome që thithen nga sipërfaqja e çelikut dhe shpërndahen në rrjetën e tij kristalore; − duke qenë një agjent reduktues aktiv, hidrogjeni atomik redukton karbidin e hekurit dhe ndërvepron me karbonin e tretur në çelik nëpërmjet reaksioneve të pakthyeshme: Fe3C + 2H2 3Fe + CH4 C + 4H CH4, duke e privuar kështu çelikun nga baza e tij forcuese. Meqenëse proceset e difuzionit, përfshirë lëvizjen e hidrogjenit, realizohen më lehtë përgjatë kufijve të kokrrave, ku mbizotërojnë pllakat e çimentitit, shkatërrimi i tyre nga hidrogjeni çon në një ndërprerje të lidhjes midis kristaliteve dhe, rrjedhimisht, në një ulje të duktilitetit të çelikut. . 30 Metani që rezulton ka një madhësi molekulare të madhe në krahasim me parametrat e rrjetës kristalore të ferritit. Si rezultat, ai nuk mund të shpërndahet nga pjesa më e madhe e metalit dhe grumbullohet në mikrokavitetet dhe defektet e tij, duke shkaktuar presion të lartë brenda zgavrës dhe duke çuar në çarje. Në këtë rast, çarjet zhvillohen përgjatë kufijve të kokrrave. Në çeliqet e butë me veti të forta të ulëta, akumulimet e metanit ndodhin kryesisht në shtresën afër sipërfaqes, duke formuar fryrje që janë qartë të dukshme në sipërfaqen e çelikut. Përveç pjesëmarrjes në formimin e metanit, hidrogjeni ka tretshmëri të mirë në metal. Përqendrimi i hidrogjenit të tretur në metal varet nga presioni i pjesshëm i hidrogjenit në ndërfaqen metal-gaz dhe mund të përcaktohet me formulën: = Ks p1/2, ku është sasia e hidrogjenit të tretur në çelik, cm3/ 100 g; p – presioni i pjesshëm i hidrogjenit, në; Ks – tretshmëria e hidrogjenit në çelik në p = 1 at. Meqenëse shpërbërja e hidrogjenit nga një metal është një proces endotermik, tretshmëria e hidrogjenit rritet me rritjen e temperaturës. Ky fenomen quhet PERMEABILITETI I HIDROGJENIT (VH) 4]. Përshkueshmëria e hidrogjenit e çelikut varet nga përmbajtja e karbonit dhe elementëve aliazh në të dhe zvogëlohet me rritjen e përmbajtjes së karbonit, siç dëshmohet nga të dhënat e dhëna në tabelë. 3. Tabela 3 Efekti i përmbajtjes së karbonit në përshkueshmërinë e hidrogjenit të çeliqeve të karbonit Metal C, wt.% VН, cm3/cm2h mm–1 Hekur teknik 0,04 5,10 Çeliku 15 0,15 4,09 Çeliku 30 0,27 2,79 Çeliku U102 nuk shfaqet. menjëherë nga momenti kur çeliku ekspozohet ndaj hidrogjenit, por pas një periudhe të caktuar kohore. Kjo kohë, gjatë së cilës nuk ka ndryshime në mikrostrukturën dhe vetitë mekanike të çelikut, quhet PERIUDHA E INKUBIMIT TË PROCESIT TË DEKARBARIMIT TË ÇELIKUT. 31 Periudha e inkubacionit ka një rëndësi të madhe praktike, pasi në thelb përcakton kohën e sigurt të funksionimit të pajisjes. Periudha e inkubacionit varet kryesisht nga temperatura dhe presioni. Me rritjen e temperaturës dhe presionit, periudha e inkubacionit zvogëlohet. Në të njëjtën kohë, kohëzgjatja e periudhës së inkubacionit ndikohet shumë nga përbërja kimike e çelikut. Temperatura dhe presioni i pjesshëm i hidrogjenit ndikojnë jo vetëm në kohëzgjatjen e periudhës së inkubacionit, por edhe në shkallën e dekarburizimit të çelikut gjatë korrozionit të hidrogjenit. 2.5.5. Korrozioni karbonil Korrozioni karbonil ndodh në proceset teknologjike që përfshijnë karbonin në presione dhe temperatura të larta. Procese të tilla përfshijnë, për shembull, prodhimin e alkooleve metil dhe butil, shndërrimin e metanit dhe monoksidit të karbonit. Në kushte normale, CO është inert ndaj metaleve. Në temperatura dhe presione të larta, monoksidi i karbonit reagon me shumë metale dhe formon karbonile. Për shembull Fe + nCO = Fe(CO)n. Hekuri dhe CO mund të formojnë tre komponime: tetrakarbonil - Fe(CO)4, pentakarbonil - Fe(CO)5 dhe nanokarbonil - Fe(CO)g. Të gjitha këto komponime janë mjaft të paqëndrueshme dhe dekompozohen kur temperatura rritet. Përbërja më e qëndrueshme midis tyre është Fe(CO)5. Formimi i karbonileve rritet me rritjen e temperaturës nga 100 ºС, pastaj, në intervalin e temperaturës 140 - 180 ºС, vërehet shpërbërja e tyre pothuajse e plotë në Fe dhe CO. Monoksidi i karbonit (II) mund të formojë komponime të ngjashme me shumë metale të tjera. Korrozioni karbonil shkakton shkatërrim dhe lirim të shtresës sipërfaqësore të metalit në një thellësi prej 5 mm. Ndryshimet në strukturën e metalit në një distancë më të madhe nga sipërfaqja nuk ndodhin më. 2.5.6. Korrozioni në jo-elektrolite JO ELEKTROLITET janë lëngje që nuk përçojnë elektricitetin. Mjete të lëngëta gërryese me origjinë inorganike - brom i lëngët, squfur i shkrirë, etj. Lëndë të lëngëta organike - benzen, kloroform etj., lëndë djegëse të lëngshme (vaj, vajguri, benzinë etj.), vajra lubrifikues. Në formën e tyre të pastër, ato janë me agresi të ulët, megjithatë, prania në to e sasive edhe të vogla të papastërtive (merkaptane14, sulfidi i hidrogjenit, uji, oksigjeni, etj.) rrit ndjeshëm aktivitetin e tyre kimik. Kështu, merkaptanët dhe sulfidi i hidrogjenit që përmbahen në naftën bruto shkaktojnë korrozion të Fe, Cu, Ni, Ag, Pb, Sn me formimin e merkaptideve të tyre (Me−(S−R)n) dhe polisulfideve (Me2Sx), përkatësisht. Gjurmët e ujit në tetraklorurin e karbonit rrisin gërryerjen e tij për shkak të formimit të përbërësve përcjellës (HCl) si rezultat i hidrolizës dhe shfaqjes së korrozionit nga mekanizmi elektrokimik: CCl4 + H2O → COCl2 + 2HCl CCl4 + 2H2O + kаt → CO2 + 4HCl. Kryesisht korrozioni i metaleve në jo-elektrolite ndodh përmes një mekanizmi kimik. Ky proces përbëhet nga disa faza, secila prej të cilave përcakton shkallën e korrozionit: 1) difuzioni i oksiduesit në sipërfaqen e metalit; 2) kimisorbimi i agjentit oksidues; 3) reaksion kimik; 4) desorbimi i produkteve të korrozionit nga sipërfaqja metalike; 5) difuzioni i produkteve të korrozionit në vëllimin e jo-elektrolitit. Në disa raste, një film mbrojtës formohet në një sipërfaqe metalike nga produktet e korrozionit, gjë që çon në frenimin e procesit të korrozionit për shkak të vështirësisë së difuzionit të oksiduesit në sipërfaqen metalike. Në varësi të vetive mbrojtëse të filmit që rezulton dhe tretshmërisë së tij, mund të instalohet difuzioni, kontrolli kinetik i difuzionit-kinetik i procesit. Temperatura ndikon ndjeshëm në shkallën e korrozionit kimik të metaleve në jo-elektrolite (shih seksionin 2.4.). Efekti i temperaturës në shpejtësinë e procesit në disa raste ndërlikohet nga ndryshimet në tretshmërinë e oksiduesit dhe filmit të produkteve të korrozionit metalik në jo-elektrolit kur temperatura ndryshon. 14 Merkaptanët janë derivate hidrosulfidë të hidrokarbureve: R – S –H. 33 Prania e ujit në jo-elektrolite aktivizon ndjeshëm efektin korroziv të papastërtive dhe shkakton korrozion elektrokimik intensiv të metaleve, d.m.th. ndryshon mekanizmi i procesit të korrozionit. Për të mbrojtur metalet në jo-elektrolite nga korrozioni kimik, zgjidhen metale dhe lidhje që janë të qëndrueshme në një mjedis të caktuar (për shembull, alumini dhe lidhjet e tij janë të qëndrueshme në benzinë e plasaritur), dhe aplikohen veshje mbrojtëse (për shembull, çeliku në një Mjedisi i sulfurit të hidrogjenit është i veshur me alumin). 2.6. Metodat për mbrojtjen e metaleve nga lloje të ndryshme të korrozionit të gazit 2.6.1. Metodat për mbrojtjen e çelikut nga korrozioni i gazit Për të mbrojtur çelikun nga gërryerja e gazit, përdoren më gjerësisht VESHJE LIGJORE rezistente ndaj nxehtësisë dhe MBROJTËSE. Lidhja - (gjermanisht legieren - "për t'u shkrirë", nga latinishtja ligare - "të lidh") - shtimi i papastërtive në përbërjen e materialeve për të ndryshuar (përmirësuar) vetitë fizike dhe/ose kimike të materialit bazë. Metodat kryesore të mbrojtjes së metaleve nga oksidimi në temperatura të larta bazohen në ALLOYING. Rezistenca e metaleve të oksiduara në temperatura të larta varet nga vetitë mbrojtëse të filmit të oksidit që mbulon metalin. Elementet që kontribuojnë në krijimin e një shtrese mbrojtëse në lidhjet hekur-karbon janë kromi, alumini dhe silikoni. Këta elementë oksidohen më lehtë në temperatura të larta në ajër sesa metali i lidhur dhe formojnë shkallë më të qëndrueshme. Sipas teorisë së lidhjes rezistente ndaj nxehtësisë, përbërësi aliazh formon një shtresë mbrojtëse në sipërfaqen e metalit që përbëhet vetëm nga oksidi i përbërësit aliazh. Elementi aliazh duhet të plotësojë këto kërkesa themelore: Oksidi i formuar në sipërfaqen e metalit duhet të jetë i vazhdueshëm, d.m.th. raporti Vok/VMe duhet të jetë >1. Shtresa okside duhet të ketë një rezistencë të lartë omike rMeO > rFe, madhësia e joneve përbërëse aliazh duhet të jetë më e vogël se madhësia e joneve të metalit bazë, d.m.th. rMe< rFe. Это условие вытекает из следующих соображений: 34 а) меньший радиус иона легирующего компонента по сравнению с ионом основного металла позволяет предполагать у легирующего компонента больший коэффициент диффузии в сплаве, что может обеспечить преимущественный выход ионов компонента на поверхность сплава и, следовательно, преимущественное образование оксида легирующего компонента; б) меньший радиус иона легирующего компонента позволит образовать оксид с соответствующими меньшими параметрами, который будет оказывать большое сопротивление диффузии через него и, следовательно, создавать большое затруднение для окисления основного металла. Это подтверждается сравнением размеров ионных радиусов железа и легирующих элементов: хрома, алюминия и кремния Ион металла Ионный радиус, Аº Fe2+ 0,74 Cr6+ 0,52 Al3+ 0,50 Si4+ 0,41. А энергия образования оксида легирующего компонента должна быть больше энергии образования оксида основного металла ΔGMeO<ΔGFeO. Если это условие не соблюдается, то оксид легирующего компонента будет восстанавливаться основным компонентом, как например, оксидная пленка меди, которая не может быть устойчивой на железном сплаве вследствие возможности протекания реакции CuO+Fe → FeO+Cu. При этом необходимо соблюдение условий: высокая температура плавления оксида легирующего компонента, низкая упругость диссоциации, а также отсутствие низкоплавких эвтектик в смеси с другими оксидами для того, чтобы оксид легирующего компонента был достаточно устойчив. В качестве примера можно привести элемент бор, который является аналогом Al по таблице Менделеева, но дает легкоплавкие оксиды. Температура плавления ВО3 − 294 0С, и поэтому бор, в отличие от алюминия, не может быть легирующим компонентом, повышающим жаростойкость. Необходимым условием является также, образование твердых растворов между легирующими компонентами и основным металлом, так как при этом условии возможно равномерное распределение этого элемента на поверхности и образование сплошной пленки оксида легирующего компонента. 35 Существует и другая теория, согласно которой легирующий элемент образует на поверхности сплава смешанные оксиды, обладающие повышенными защитными свойствами, по сравнению с оксидами чистых компонентов. Механизм повышения жаростойкости можно свести к тому, что легирующий компонент должен уменьшить возможность образования в окалине вюститной фазы (FeO) на стали и, таким образом, благоприятствовать образованию шпинельной фазы типа Fe3O4ּγFe2O3 с возможно меньшим параметром решетки. Жаростойкие легированные стали имеют на поверхности оксидные слои со структурой именно шпинели. Еще более высокими защитными свойствами обладают сложные шпинели типа FeOּMe2O3 или Fe2O3 ּMeO. С целью экономии легированных сталей жаростойкость углеродистых сталей повышают, применяя защитные покрытия. Для нанесения на сталь защитных покрытий пользуются следующими методами: ТЕРМОДИФФУЗИОННЫМ, НАПЛАВКОЙ И ПЛАКИРОВАНИЕМ. Для нанесения покрытий термодиффузионным методом стальное изделие после очистки поверхности механическим способом и травлением помещают в реакционную смесь, состоящую из порошка наносимого металла и катализатора. Например, при нанесении алюминиевых покрытий смесь содержит измельченный ферроалюминиевый и хлористый аммоний. Процесс насыщения поверхности стали алюминием (АЛИТИРОВАНИЕ) производится при высоких температурах (900−9500С), в результате чего происходит разложение хлористого аммония на аммиак и хлористый водород: NH4Cl → NH3 + HCl, который, в свою очередь, взаимодействует с алюминием 2Al +6HCl → 3H2 + 2AlCl3. На поверхности стального изделия происходит выделение атомарного алюминия AlCl3 +Fe → FeCl3 + Al, который диффундирует в сталь. Значительное повышение жаростойкости стали термодиффузионным покрытием обусловлено образованием окислов Al2O3 или двойных окислов FeAl2O4 в алитированном слое. На поверхности хромированных или силицированных изделий образуются соответствующие оксиды хрома или кремния. 36 НАПЛАВКА − это нанесение слоя металла или сплава на поверх- ность изделия посредством сварки плавлением. ПЛАКИРОВА́НИЕ − (фр. plaquer − накладывать, покрывать) термомеханическое покрытие − нанесение на поверхность металлических листов, плит, проволоки, труб тонкого слоя другого металла или сплава термомеханическим способом. Для защиты от карбонильной коррозии применяют хромистые стали с содержанием 30 % Сг, хромоникелевые стали с содержанием 23 % Сг и 20 % № и марганцевые бронзы доя работы при температуре до 700 °С и давлении до 35 МПа. При более низких параметрах возможно применение менее легированных сталей типа Х18Н9. Сырьем для синтеза мочевины CO(NH2)2 является NH3 и СО2. Процесс протекает при температуре 175−190°С и давлении 20 МПа. Хромистые нержавеющие стали различных марок непригодны для изготовления основных аппаратов. Наибольшую стойкость имеют стали, легированные молибденом, и хромникельмолибденовомедные стали. Важным фактором для повышения коррозионной устойчивости является тщательная очистка газов от сероводорода и дополнительное введение в систему кислорода в количестве 0,5- 1,0 об.% от содержания СО2. При сернистой коррозии сера и ее соединения - сернистый ангидрид (SO2), сероводород (H2S), меркаптаны или тиоспирты и т.д. являются достаточно агрессивными, коррозионно−активными веществами. Наиболее активным компонентом при высокотемпературной газовой коррозии является сероводород. Он даже более опасен, чем диоксид серы. Сернистый газ SO2 является исходным продуктом при производстве серной кислоты. Его получают при обжиге серного колчедана, сжигании серы, из сероводорода при утилизации отходящих газов металлургических производств. Чугунные детали скребков конверторных печей кипящего слоя, зубья и гребки колчеданных печей, котлы−утилизаторы, сухие электрофильтры, газоходы обжиговых газов в производстве серной кислоты часто выходят из строя вследствие газовой коррозии. В результате коррозии черных металлов в сернистом газе при температурах 300°С и выше образуется слоистая окалина, состоящая из FeS, FeO и др. 37 При температуре газа более 400 °С для деталей из чугуна характерно увеличение объема металла, достигающего 10 % от начальной величины. При этом резко снижается прочность материала. Детали испытывают коробление, трескаются и разрушаются. Это явление называется «ростом» чугуна и объясняется внутренним окислением металла. Максимальный рост чугуна наблюдается при 700 °С. К ростоустойчивым чугунам относятся высоколегированные хромистые чугуны, карбидный чугун типа «пирофераль» и «чугаль» Сернистый газ при высоких температурах окисляет никель. При этом образуется окалина, в состав которой входят NiS и NiO: 3Ni + S02 = NiS + 2NiO. Сернистый никель образуется и при действии на металл сероводорода: Ni + H2S = NiS + Н2. Сульфид никеля с металлическим никелем образует легкоплавкую эвтектику с температурой плавления около 625 °С. Образование этой эвтектики в сталях, содержащих никель, происходит преимущественно по границам зерен, вызывая разрушение металла. Стали с содержанием никеля выше 15 % очень чувствительны к действию сернистого газа. В процессе окисления они теряют механическую прочность. Поэтому при работе с газовой средой, содержащей диоксид серы, при температурах до 400 °С используют углеродистые стали, а при более высоких температурах − хромистые стали. Наиболее употребительны жаростойкие стали − 4Х9СА, Х6СЮ, XI7, ОХ 17 Г, X1800, Х25Т. Интенсивное образование окалины происходит при температурах выше 800−1000 °С. К жаропрочным сталям в этой среде относятся Х5М, Х6СМ, XI8Н12Т, Х23Н18. Рабочая температура для этих сплавов 550−600 °С (для Х23Н18 − 1000 °С). Сухой сернистый газ реагирует с алюминием очень медленно. Поэтому алюминий используют для защиты от коррозии деталей и узлов теплообменников и контактных аппаратов. Сухой сероводород при комнатной температуре не представляет опасности для обычных углеродистых сталей. С повышением температуры опасность сероводородной коррозии углеродистых сталей значительно увеличивается. При температуре выше 300 °С 38 железо подвергается сильной коррозии в серосодержащих газовых средах. Легирование хромом в количестве > 12% rrit rezistencën ndaj korrozionit në temperatura deri në 700−800 °C. Kur çeliqet e kromit gërryen, formohet peshore, shtresa e jashtme e së cilës përbëhet nga sulfur hekuri. Praktikisht nuk ka krom në këtë shtresë. I gjithë kromi i oksiduar është i përqendruar në shtresën e brendshme, e cila ka një veti mbrojtëse. Ferriti Fig. 5 10 15 20 ka rezistencë të mirë kimike në një atmosferë squfuri-Cr, wt.% hidrogjen. 6. Shkalla e korrozionit të lidhjeve të kromit që përmbajnë 25–30% çelik të ngurtë në avull vaji në 650 °C. Numrat në kthesa tregojnë krom. Përmbajtja e H2S, % Veçanërisht e rrezikshme është prania e kombinuar e përbërjeve të squfurit dhe përbërësve të tjerë gërryes. Kështu, në industrinë e naftës, gjatë përpunimit termik të vajrave sulfurorë, një rrezik i veçantë përbën një përzierje e sulfurit të hidrogjenit dhe hidrogjenit. Nga ato të paraqitura në Fig. 6 të dhënat tregojnë se shkalla e korrozionit të çeliqeve të kromit rritet me rritjen e përqendrimit të sulfurit të hidrogjenit në avujt e vajit. Në këtë rast, një rritje në përqendrimin e H2S me 10 herë shkakton një rritje të shkallës së korrozionit me më shumë se 12-15 herë. Kur karburanti digjet, formohen përzierje komplekse gazi që përmbajnë O2 dhe okside të ndryshme, duke përfshirë papastërtitë e squfurit. Në këto raste vërehet korrozioni SULFIDE-OXID. Filmi mbrojtës në metal zakonisht përbëhet nga disa shtresa. Shtresa e jashtme është e pasuruar me oksigjen dhe përbëhet nga oksid metali, dhe shtresat e brendshme ngjitur me sipërfaqen e metalit përmbajnë sasi të shtuara të squfurit dhe sulfureve. Nëse djegia e karburantit prodhon hirin, i cili përmban oksid vanadiumi V2O5, atëherë shkalla e korrozionit rritet shumë shpejt. Shkaqet e korrozionit vanadium të çeliqeve u diskutuan më herët (shih seksionin 2.5.2). 39 Çeliqet e kromit që përmbajnë 4−6% Cr konsiderohen gjysmë rezistente ndaj nxehtësisë. Çeliqet e kësaj klase, për shkak të disponueshmërisë së tyre, rritjes së rezistencës ndaj korrozionit dhe forcës, përdoren gjerësisht në industrinë e naftës për prodhimin e njësive të plasaritjes. Rezistenca ndaj nxehtësisë e këtyre çeliqeve në ajër dhe në gazrat e gripit me një përmbajtje të konsiderueshme të përbërjeve të squfurit në temperaturat 500−600 ºС është afërsisht 3 herë më e lartë se rezistenca ndaj nxehtësisë e çeliqeve të palidhur. 2.6.2. Metodat e mbrojtjes nga korrozioni hidrogjenik Një nga mënyrat kryesore për të rritur rezistencën ndaj hidrogjenit të çeliqeve është aliazhimi i tyre me elementë të fortë karabitformues, të cilët formojnë karbide më të qëndrueshme se çimentiti. Elementë të tillë janë kromi, tungsteni, molibden, vanadium, niobium, titani. Lloji i fazës së karbitit ka një ndikim të madh në rezistencën ndaj hidrogjenit të çelikut. Në çeliqet e lidhur me krom, rezistenca ndaj hidrogjenit zvogëlohet në varësi të përbërjes së fazës karabit në seritë e mëposhtme: (Cr,Fe)23C6(Cr,Fe)23C6 + (Cr,Fe)7C3(Cr,Fe) 7C3 (Cr,Fe)7C3 + (Fe,Cr)3C(Fe,Cr)3C Seria e mësipërme tregon se rezistenca më e madhe ndaj hidrogjenit në çeliqet e kromit arrihet me formimin e karbiteve të (Cr,Fe ) Lloji 23C6. Formimi i këtij lloji karabit me përmbajtje karboni në çelik prej 0,05% ndodh kur në çelik ka të paktën 6% krom, dhe me rritjen e përmbajtjes së karbonit është e nevojshme një rritje e përmbajtjes së kromit. Kur aliazhoni çelikun me elementë më të fortë që formojnë karabit sesa kromi, rezistenca më e madhe ndaj hidrogjenit e çelikut arrihet kur këta elementë përmbahen në sasi të mjaftueshme për të lidhur të gjithë karbonin në karbide të tipit MeC. Në rastet kur elementët e pajisjeve funksionojnë në kushte të niveleve të larta të ngarkesave mekanike, çeliqet rezistente ndaj hidrogjenit duhet gjithashtu të kenë rezistencë ndaj nxehtësisë. Për këto kushte, përdoren çeliqet me 3% Cr, të lidhur me Mo, V ose W, për shembull çeliku i klasës 20Х3МВФ (EI579). Notat e çelikut X5M, X5VF, X9M, 1X13 kanë rezistencë edhe më të madhe. Në kushtet më të rënda, përdoren çeliqe austetike krom-nikel rezistente ndaj nxehtësisë 0Х18Н10Т ose Х17Н17М2Т. 40 Një metodë tjetër e përdorur gjerësisht për rritjen e rezistencës ndaj hidrogjenit të çeliqeve është MBUSHJA e tyre me metale që kanë një përshkueshmëri më të ulët të hidrogjenit se metali bazë. Kjo bën të mundur uljen e përqendrimit të hidrogjenit në ndërfaqen midis metalit bazë dhe shtresës së veshjes dhe zvogëlimin e efektit të tij agresiv në metalin bazë. Llogaritja e presionit efektiv të hidrogjenit në ndërfaqen metalike bazë-veshje kryhet sipas ekuacionit: 1/2 1/2 p2 = p1 (11) + ku p1 është presioni i hidrogjenit në sipërfaqen e jashtme të shtresa e veshjes; p2 është presioni efektiv i hidrogjenit në ndërfaqen shtresë-metal bazë; = VН1/VН2 është raporti i përshkueshmërisë hidrogjenore të shtresës së veshjes dhe metalit bazë, përkatësisht; = l1/l2 – raporti i trashësisë së shtresës së veshjes dhe metalit bazë, përkatësisht. Me qëllim të rritjes së përshkueshmërisë së hidrogjenit, metalet dhe lidhjet mund të vendosen në një rresht: alumini, bakri, nikel, X18N10T, 0X13. Në praktikë, industria kimike dhe e rafinimit të naftës përdorin kryesisht bimetale nga karboni ose çeliqe me aliazh të ulët me një shtresë mbrojtëse prej çeliku 0Х13 ose Х18Н10Т. Temperatura e funksionimit të pajisjeve të bëra nga çeliku i zgjedhur duhet të mbahet 25°C nën atë në të cilën është i mundur korrozioni hidrogjenor. Kapitulli 3. KORROZIONI ELEKTROKIMIK I METALEVE 3.1. Potencialet e elektrodeve të metaleve Kur një metal zhytet në një elektrolit, në kufirin e fazës ndodh një kërcim potencial për shkak të formimit të një shtrese elektrike të dyfishtë 5, 6. Nëse një pllakë metalike, për shembull, një pllakë bakri, zhytet në ujë ose në një zgjidhje kripe bakri, atëherë jonet Cu2+ të ngarkuar pozitivisht do të fillojnë të lëvizin nga shtresa metalike e vendosur në kufirin me ujin në ujë. Në këtë rast, një tepricë e elektroneve shfaqet në rrjetën kristalore të metalit, dhe pllaka fiton një ngarkesë negative. Një tërheqje elektrostatike lind midis pllakës së ngarkuar negativisht dhe joneve pozitive që kanë kaluar në tretësirë, gjë që parandalon kalimin e mëtejshëm të joneve të bakrit në tretësirë, d.m.th., procesi i tretjes së metalit ndalon. Në të njëjtën kohë, zhvillohet procesi i kundërt: jonet e bakrit nga tretësira, duke iu afruar sipërfaqes së pllakës, pranojnë elektrone prej saj dhe kalojnë në një gjendje neutrale. Pas një periudhe të caktuar kohe, krijohet një gjendje e ekuilibrit dinamik, në të cilin shpejtësia e kalimit të joneve nga metali në tretësirë është e barabartë me shpejtësinë e shkarkimit të joneve nga tretësira në metal. Fenomeni i përshkruar është paraqitur në mënyrë skematike në Fig. 7 (jonet metalike tregohen të pahidratuara për thjeshtësi). Ekuilibri ndërmjet joneve në tretësirë dhe metalit për shembullin në shqyrtim shprehet me ekuacionin: Cu2+р−р + 2ē Cuºkristal Në ekuacionin e ekuilibrit të një reaksioni elektrokimik, elektronet zakonisht shkruhen në anën e majtë. d.m.th., procesi i reduktimit është shkruar. Fig.7. Skema e paraqitjes së një shtrese elektrike të dyfishtë në ndërfaqen metal-tretësirë Siç mund të shihet nga shembulli i mësipërm, kur një metal bie në kontakt me një tretësirë të kripës së tij, dy fazat kontaktuese fitojnë ngarkesa të kundërta. Si rezultat, një shtresë elektrike e dyfishtë formohet në ndërfaqen e fazës dhe një kërcim potencial () ndodh midis metalit dhe tretësirës. Nëse dy pllaka metalike të metaleve të ndryshme (M1 dhe M11) ulen në tretësirë, do të lindë një çift galvanik në të cilin metali më pak aktiv do të reduktohet dhe ai më aktivi do të oksidohet (Fig. 8). Oriz. 8. Skema e funksionimit të një qelize galvanike Në përgjithësi, puna e këtij çifti galvanik përcaktohet nga diferenca potenciale (emf e qelizës galvanike) dhe shoqërohet me balancën e gjysmëreaksioneve: E = ox − red, ku ox është potenciali i oksiduesit, V; red – potencial agjenti reduktues, V. Burra+(ox) + ne ⇆ Меº Meº(kuqe) − ne ⇄ Меn+. Kur numri i kationeve që kalojnë në tretësirë për njësi të kohës bëhet i barabartë me numrin e kationeve të depozituara në sipërfaqen e metalit, do të ndodhë ekuilibri dinamik dhe procesi i tretjes do të ndalet. Prandaj, kalimi i një numri të madh të atomeve të joneve metalike në zgjidhje në kushte të tilla është i pamundur. Sidoqoftë, nëse ekuilibri i shtresës së dyfishtë elektrike prishet nga shkarkimi i elektroneve ose heqja e atomeve të joneve të metalit, procesi i korrozionit do të vazhdojë pa pengesa. Potencialet e elektrodës së metaleve që janë në ekuilibër me jonet e tyre quhen ekuilibër. Vlera e potencialit të ekuilibrit mund të llogaritet për çdo aktivitet jonik duke përdorur ekuacionin Nernst: E = Eº + (RT /nF)ln(aMen+), (12) n+ ku Eº është diferenca standarde e potencialit në aMe = 1; R – konstante universale e gazit, 8,3 kJ/kmolK; T – temperatura absolute, ºK; n – valencë metalike; F – Numri Faraday 96500 C/mol; 43 aМen+ – aktiviteti i joneve metalike në mol/l. Nëse zëvendësojmë të gjitha konstantat në 25°C (T = 298 K) dhe shumëzojmë me 2.3 për të kaluar nga logaritmet natyrore në ato dhjetore, marrim shprehjen e mëposhtme E = Eº + (0.0592/n)log(aMen+), ( 13) Kur tretësira hollohet, potenciali i metalit zhvendoset në anën negative. Nëse, për shembull, aktiviteti i joneve Zn2+ në një tretësirë të kripës së zinkut është 10–2 mol/l, atëherë potenciali ekuilibër i zinkut të zhytur në këtë tretësirë do të jetë i barabartë me: E = - 0,76 + (0,0592/2) lg10−2 = − 0,819 V, (14) Me një rritje të përqendrimit të joneve metalike në tretësirë, potenciali metalik, përkundrazi, zhvendoset në drejtim pozitiv. Kështu, nëse aktiviteti i joneve të zinkut merret si 10 mol/l, atëherë potenciali i zinkut do të jetë E = − 0,76 + (0,0592/2)log10 = − 0,73 V, (15) Potencialet e elektrodave të metaleve në të cilat procesi i shkëmbimit që përcakton potencialin përfshin jo vetëm të tijin, por edhe jone dhe atome të tjera, të cilat quhen jo ekuilibër ose të pakthyeshëm. Për potencialet jo ekuilibër, formula Nernst nuk është e zbatueshme, pasi reaksionet që ndodhin në metal, d.m.th. humbja dhe përvetësimi i elektroneve ndodhin në mënyra të ndryshme dhe potenciali nuk mund të karakterizojë fillimin e ekuilibrit të një reaksioni të vetëm në elektrodë. Madhësia e potencialeve jo ekuilibër ndikohet nga natyra e elektrolitit, temperatura, lëvizja e elektrolitit, përqendrimi i tretësirës, etj. Për shembull, potenciali i aluminit në 3% NaCl është − 0,6 V, në 0,05 M Na2SO4 − 0,47 V, dhe në 0,05 M Na2SO4 + H2S − 0,23 V. Për shkak të heterogjenitetit elektrokimik të sipërfaqes së metalit ose elektrolitit, në metal formohen zona me potenciale të ndryshme. Arsyet kryesore që shkaktojnë heterogjenitetin elektrokimik të një sipërfaqeje metalike mund të jenë si më poshtë: − LIKACIONI − (La liquation, Saigerung) - është aftësia e lidhjeve për t'u shpërbërë gjatë kalimit nga gjendja e lëngët në të ngurtë në përbërës ose komponime individuale që kanë pika të ndryshme shkrirjeje. ; − heterogjeniteti i filmit mbrojtës; 44 – heterogjeniteti i kushteve fizike – temperatura e pabarabartë në zona të ndryshme të metalit, prania e zonave të deformuara, përqendruesit e stresit; − heterogjeniteti i elektrolitit - dallimi në përqendrimin e oksigjenit ose në përqendrimin e kripës, vlera e ndryshme pH. Zonat me potencial më negativ quhen ANODIK, zonat me potencial më pozitiv quhen KATOdë. Shkalla e heterogjenitetit të sipërfaqes së metalit përcaktohet nga ndryshimi në potencialet e seksioneve katodë dhe anodike. 3.2. Termodinamika e korrozionit elektrokimik Mundësia e shfaqjes spontane të procesit të korrozionit përcaktohet nga zvogëlimi i energjisë së lirë (potenciali izobarik-izotermik) G< 0: G = −nFE < 0, (16) где n – эквивалентное число электронов; Е = К – А – эдс гальванического элемента, образующегося при контакте металла с окислителем в процессе коррозии; К – обратимый потенциал катодной реакции (окислителя); А – обратимый потенциал анодной реакции (металла – восстановителя); F – число Фарадея. Принципиальная возможность протекания процесса электрохимической коррозии металла имеет место при К > A ose E > 0. Potenciali redoks i kthyeshëm i agjentit oksidues në elektrolit duhet të jetë më pozitiv në këto kushte sesa potenciali i kthyeshëm i metalit në të njëjtat kushte. Për të vendosur kufijtë e mundësisë termodinamike të korrozionit elektrokimik të metaleve, mund të përdoren diagramet Pourbaix. Diagramet janë paraqitje grafike të varësisë së potencialeve të elektrodave të kthyeshme (në volt në shkallën e hidrogjenit) nga pH e tretësirës për gjendjet e ekuilibrit: me pjesëmarrjen e elektroneve - vija horizontale; me pjesëmarrjen e elektroneve dhe joneve H+ ose OH– - vija të pjerrëta; me pjesëmarrjen e joneve H+ dhe OH–, por pa pjesëmarrjen e elektroneve (vlera e pH e formimit të hidratit) – vija vertikale. 45 Fig.9. Diagrami E – pH për sistemin Al-H2O në 25°C Si shembull në Fig. Figura 9 tregon diagramin Pourbaix për sistemin Al – H2O. Çdo rajon i diagramit korrespondon me një gjendje termodinamikisht të qëndrueshme: gjendjen metalike, në formën e joneve në tretësirë, në formën e oksideve ose hidroksideve. Meqenëse vendosja e ekuilibrit në një zgjidhje varet jo vetëm nga jonet e hidrogjenit, por edhe nga jonet e tjera, përcaktimi i rajoneve formohet nga disa linja ekuilibri. Secila prej këtyre linjave korrespondon me një aktivitet specifik të joneve përkatëse. Në rajonin në fund të diagramit, metali i aluminit është termodinamikisht i qëndrueshëm dhe nuk gërryhet. Në pjesën e majtë mbi vijën horizontale, gjendja e aluminit në formë të joneve Al3+ në tretësirë është termodinamikisht e qëndrueshme. Zona e mesme korrespondon me qëndrueshmërinë e fazave të ngurta të oksidit Al2O3 ose hidroksidit Al(OH)3, të cilat formojnë një film mbrojtës në sipërfaqen e aluminit gjatë procesit të korrozionit. Ana e djathtë karakterizon kushtet në të cilat alumini do t'i nënshtrohet korrozionit me formimin e anionit AlO2– në tretësirë. 3.3. Mekanizmi i korrozionit elektrokimik Korrozioni i metaleve në elektrolite ndodh me formimin e çifteve galvanike, të cilat quhen çifte korrozioni. Nëse ulni përkatësisht pllakat e zinkut dhe hekurit në një enë me tretësira të zinkut dhe klorureve të hekurit (të ngjashme me diagramin e pH me në Fig. 8) dhe i lidhni ato me një përcjellës të jashtëm, atëherë një miliammetër i lidhur me qark do të tregojë prania e rrymës elektrike në qark. Formohet një çift galvanik në të cilin zinku do të oksidohet dhe do të hyjë në tretësirë (anodë). Hekuri nuk do të oksidohet dhe nuk do të hyjë në tretësirë, pasi, duke u kombinuar me zink, ai vepron si një katodë. Hekuri, i pa kombinuar me zink, gërryhet në një zgjidhje të ngjashme. Në praktikë, lidhjet përmbajnë një numër të madh mikroelementesh dhe metale të ndryshme. Me fjalë të tjera, metalet e ndryshme ndodhen në të njëjtin rrafsh në kontaktin e tyre të drejtpërdrejtë me njëri-tjetrin dhe me elektrolitin. Rezultati është një qelizë multielektrodike në të cilën anodet alternojnë me katoda. Në vendet anodike, jonet metalike (Men+) kalojnë në tretësirë. Elektronet e lëshuara (nē) lëvizin nëpër metal nga anoda në katodë. Tretja e joneve metalike në tretësirat ujore duhet të përfaqësohet si rezultat i bashkëveprimit të joneve - atomeve të vendosura në sipërfaqen e jashtme të metalit me molekulat polare të ujit. Ndërveprimi i joneve të metalit të tretur me dipolet e ujit shkaktohet nga forcat e tërheqjes elektrostatike. Si rezultat, rreth çdo joni, në një masë më të madhe ose më të vogël (në varësi të madhësisë së ngarkesës së tij), formohet një guaskë dipolesh uji (fenomeni i hidratimit). Për shkak të hidratimit, rrezja efektive e jonit rritet, si rezultat i së cilës lëvizshmëria e joneve të hidratuar zvogëlohet ndjeshëm. Procesi i hidratimit shoqërohet me çlirimin e energjisë, ndërsa procesi i dehidrimit kërkon energji. Përveç dipoleve të ujit, joni mund të mbulohet me një guaskë dipolesh të tjera. Në përgjithësi, ky fenomen quhet zgjidhje15. Kështu, korrozioni elektrokimik përbëhet nga këto faza, që ndodhin paralelisht: - një proces anodik, i cili konsiston në kalimin e joneve metalike nga sipërfaqja në tretësirë dhe hidratimi i tyre: ZGJIDHJA - procesi i formimit të lidhjeve midis një tretësi dhe një tretësish të tretur. substancë, produkti i solvacionit është SOLVATE. Nëse tretësi është uji, atëherë HIDRATIMI, produkti i hidratimit është HIDRATI. 15 47 Meº − nē Meshkuj+ + mH2O Burra+ mH2O. hidrati është një proces katodik, i cili konsiston në asimilimin (kapjen) e elektroneve nga ndonjë depolarizer (D): D + nē . Meqenëse proceset anodike dhe katodike janë të pavarura dhe ndodhin më lehtë: anodiku në zonat me potencial fillestar më negativ të sipërfaqes, dhe katodik në ato më pozitivë - dhe në kushte praktike heterogjeniteti elektrokimik i sipërfaqes së metalit është i nevojshëm për këtë. vihet re, këto procese ndodhin kryesisht në një mënyrë të lokalizuar. Rrjedha e rrymës elektrike kryhet në metal - me lëvizjen e elektroneve nga zonat anodike në katodë, dhe në tretësirë, në të njëjtën kohë, ndodh lëvizja e joneve. Kështu, forca aktuale mund dhe shërben si një kriter për shkallën e korrozionit elektrokimik. Llogaritni konsumin e materialit të anodës, d.m.th. sasia e metalit të gërryer nga 1 cm2 e sipërfaqes së tij mund të përdoret duke përdorur formulën e Faradeit: K = Q A / F n = i A / F n, ku K është sasia e gërryerjes. metal, g/cm2; Q është sasia e energjisë elektrike që rrjedh gjatë kohës τ, [s] ndërmjet seksioneve të anodës dhe katodës; i – dendësia e rrymës, A/cm2; F – numri i Faradeit; n – valencë metalike; A është masa atomike e metalit. 3.4. Polarizimi i proceseve të elektrodës dhe shkaqet e tij Kur elektrodat e një elementi korrozioni qarkullojnë shkurt, ndodh një ndryshim i rëndësishëm në potencialet e elektrodës, por rezistenca e çiftit praktikisht nuk ndryshon. Në këtë rast, vërehet një ulje e forcës elektromotore të elementit të korrozionit. Një ndryshim në potencialet fillestare, që çon në një ulje të rrymës së korrozionit, dhe, rrjedhimisht, në shkallën e korrozionit, quhet POLARIZIM. Ndryshimi i potencialeve (16) të elektrodave gjatë funksionimit të elementit të korrozionit tregon se potenciali katodik bëhet më negativ (POLARIZIMI KATODIK), kurse potenciali i anodës bëhet më pozitiv (POLARIZIMI ANODIK): K = Ko – K, (17) o A = A + A, (18) ku K dhe A janë vlerat potenciale të elektrodave të elementit punues; Кº dhe Аo – vlerat fillestare të potencialeve të elektrodës para mbylljes së qarkut; K dhe A – zhvendosja (polarizimi) potencial i katodës dhe anodës. Kështu, diferenca e potencialit E = K − A zvogëlohet. POLARIZIMI është pasojë e vonesës së proceseve të elektrodës nga rrjedha e elektroneve në një element me qark të shkurtër. Rezistenca omike (R) ka pak efekt në reduktimin e rrymës së korrozionit, pasi ato zakonisht janë të vogla. Rezistencat e polarizimit janë të një rëndësie të madhe, të cilat shoqërohen ose me vështirësi në shkarkimin e elektroneve në katodë (Pk), ose me vështirësi në kalimin e joneve pozitive të metalit nga rrjeta metalike në tretësirë (PA). Këto rezistenca, të cilat kanë një dimension omik, janë shkaku kryesor i uljes së shkallës së korrozionit elektrokimik. Forca e rrymës në momentin e mbylljes së qarkut në përputhje me ligjin e Ohm-it përcaktohet me formulën Iinit = (Кº − Аo)/R, (19). Vlera fillestare e rrymës së korrozionit (Iinit) është drejtpërdrejt proporcionale me ndryshimin midis vlerave fillestare të potencialit Kº dhe Ao, me kusht që Kº > Ao, dhe në përpjesëtim të zhdrejtë me rezistencën. Rryma e korrozionit në gjendje të qëndrueshme (qeliza e punës) përcaktohet nga ekuacioni: Qeliza e punës = (K – A)/(R + PA + PK). Kështu, unë punoj. el−ta< Iнач. Снижению скорости электрохимической коррозии или уменьшению коррозионного тока могут способствовать: − уменьшение степени термодинамической стабильности, т. е. сближение равновесных потенциалов анодного Аº и катодного Кº процессов; − торможение катодных процессов, иначе говоря, увеличение катодной поляризуемости РК; 49 − торможение анодных процессов, то есть увеличение анодной поляризуемости РА. Поляризация является весьма желательным явлением в коррозионных процессах, так как электроды замкнуты накоротко, омическое сопротивление их невелико и, следовательно, не будь поляризационных сопротивлений, коррозия протекла бы весьма интенсивно. Всякое воздействие, уменьшающее поляризацию, увеличивает скорость коррозионных процессов. Факторы, уменьшающие поляризацию электродов коррозионного элемента, называют ДЕПОЛЯРИЗАТОРАМИ, а процессы, протекающие при этом – АНОДНОЙ И КАТОДНОЙ ДЕПОЛЯРИЗАЦИЕЙ. 3.4.1. Анодная поляризация Анодная поляризация обычно оказывает меньшее влияние на изменение разности потенциалов элемента по сравнению с катодной поляризацией. Анодный процесс, заключающийся в ионизации металла и гидратировании ионов по уравнению: Men+ + mH2O → Men+ ∙ mH2O, может протекать беспрепятственно только при условии беспрерывного отвода образующихся ионов из прианодной зоны. Торможение анодного процесса будет иметь место и в случае отставания скорости выхода ионов металла в раствор от скорости перетекания электронов на катодные участки. В результате анодной поляризации отрицательный заряд на металлической обкладке двойного слоя уменьшается, а следовательно, потенциал металла сдвинется в положительную сторону (или станет менее отрицательны). Таким образом, анодная поляризация происходит или в связи с тем, что скорость анодного процесса не поспевает за скоростью отвода электронов (перенапряжение ионизации металла), или потому, что из−за недостаточно быстрого отвода перешедших в раствор ионов металла повышается концентрация этих ионов в прианодной зоне (концентрационная поляризация). Чем легче протекает анодный процесс, тем меньше его поляризуемость. Следить за тем, легко ли протекает анодный процесс или он заметно тормозится, можно по ходу поляризационной анодной кри50 Потенциал, В вой. Эта кривая показывает зависимость потенциала электрода от плотности анодного тока на нем. Такая кривая показана на рис. 10. Удаление продуктов анодной реакции вызывает деполяризацию анода. Деполяризация анода может быть ускорена перемешиЕ Е ванием раствора, образованием о А таких продуктов коррозии, которые выпадают в осадок, а также путем образования комплексных соединений. Вследствие связывания в комплекс ионов металла, перешедших в раствор, концентрация их в прианодной зоне уменьшаПлотность тока, мА/см2 ется; анодная поляризация сниРис. 10. Анодная поляризаци- жается и переход ионов в расонная кривая твор, т.е. коррозия, усиливается. Например, медь и ее сплавы образуют в растворе аммиака хорошо растворимый комплексный ион 2+. Этим объясняется сильная коррозия меди и ее сплавов в аммиачных растворах. Поляризация анодного процесса в обычных условиях происходит в незначительной степени. Исключение представляет случай возникновения анодной пассивности. 3.4.2. Катодная поляризация Протекание катодной реакции могут тормозить два основных процесса: − торможение за счет трудности протекания самой реакции присоединения электрона деполяризатором, т. е. задержка в реакции nē + D (Dnē). Затруднение протекания катодного процесса по этой причине называется ПЕРЕНАПРЯЖЕНИЕМ РЕАКЦИИ КАТОДНОЙ ДЕПОЛЯРИЗАЦИИ; − торможение в процессе подвода к катодной поверхности деполяризатора (D) или отвода от катодной поверхности продуктов восстановления деполяризатора (Dnē). Затруднение катодного процесса по этой причине называется КОНЦЕНТРАЦИОННОЙ ПОЛЯРИЗАЦИЕЙ КАТОДА. 51 Всякий процесс захвата электрона на катоде, т.е. всякий процесс восстановления какого−либо вещества может являться катодной деполяризующей реакцией. Деполяризация катода может протекать различными путями: − ассимиляция электронов ионами: 2H+ +2ē H2, Fe3+ + ē Fe2+, Cu2+ + ē Cu+, S2O8– + ē 2SO42–; − ассимиляция электронов нейтральными молекулами: О2 + 4ē + 2Н2О 4ОН–, Cl2 + 2ē 2Cl–, Н2О2 + 2ē 2ОН–; − ассимиляция электронов нерастворимыми пленками Fe3O4 + 2ē + H2O 3FeO + 2OH–, Fe(OH)3 + ē Fe(OH)2 + OH–; − ассимиляция электронов органическими соединениями RO + 2ē + 4H+ RH2 + H2O, R + 2ē + 2H+ RH2, где R – радикал или органическая молекула. Наиболее часто встречаемыми и наиболее важными катодными деполяризационными процессами являются два: а) разряд и выделение водорода (деполяризатор ион водорода) 2H+ +2ē H2. В этом случае коррозионный процесс называется коррозией с ВОДОРОДНОЙ ДЕПОЛЯРИЗАЦИЕЙ. Этому виду коррозии подвержены электроотрицательные металлы в кислотах и весьма электроотрицательные металлы, например Mg, в воде и нейтральных растворах электролитов; б) восстановление атомов или молекул кислорода электронами, притекающими на катодных участках − КИСЛОРОДНАЯ ДЕПОЛЯРИЗАЦИЯ: О2 + Н2О + 4ē 4ОН–. Деполяризатором здесь является растворенный в электролите кислород. Этот вид коррозии является наиболее распространенным. Так корродирует большинство металлов в атмосфере, почве, нейтральных растворах электролитов. Иногда оба деполяризующие процесса протекают параллельно, как например, при коррозии железа в разбавленных растворах серной кислоты в присутствии кислорода воздуха. 52 3.4.3. Водородная деполяризация Протекание коррозии металла с водородной деполяризацией возможно, если (Me)обр < (н2)обр. (20) Согласно уравнению Нернста (н2)обр = (н2)ºобр + (RT2,3/F)lg(aH+/pH21/2), (21) о где (н2) обр – стандартный обратимый потенциал водородного электрода при всех температурах (при ан+ = 1 и рН2 = 1 ат); R – универсальная газовая постоянная. Ниже приведены значения обратимого потенциала водородного электрода (н2)обр при 25 оС и различных значениях рН среды и pН2. рн2, ат 5∙10 1 –7 0 + 0,186 0 рН среды 7 − 0,228 − 0,414 14 − 0,641 − 0,828 Изменение потенциала катода при катодной деполяризации выделением водорода подчиняется ближе всего логарифмической зависимости от плотности тока. Эта зависимость определяется выражением: К = Кº – (а + b lg i). (22) Для очень малых плотностей катодного тока (меньше 10–4 – 10–5 А/см2) потенциал от плотности тока имеет линейную зависимость. Поэтому при i = 0 К = Кº, а не бесконечности, как это следует из уравнения. Поляризуемость катода при выделении на нем водорода принято определять ВЕЛИЧИНОЙ ПЕРЕНАПРЯЖЕНИЯ ВОДОРОДА на данном материале катода. Под ПЕРЕНАПРЯЖЕНИЕМ ВОДОРОДА понимают сдвиг потенциала катода при данной плотности тока в отрицательную сторону по сравнению с потенциалом водорода в том же растворе (без наложения тока) и обозначают через η η = − (К − Кº) (23) или η = a + blgI, (24) где b – константа, не зависящая от материала, рассчитывается как 2,3 ∙ 2RT / nF; а − константа, зависит от природы и состояния поверхности материала катода и численно равна величине перенапряжения при плотности тока, равной единице. 53 Поляризационная кривая для случая водородной деполяризации имеет вид кривой Кº MN (рис. 11). − N 1 M η Рис. 11. Катодная поляризационная кривая для процесса водородной деполяризации ко или (н2)обр i1 0 i P Если по оси абсцисс взять не плотность тока, а логарифм этой величины, то кривая перенапряжения будет прямой. В этом случае константа b является тангенсом угла наклона полученной прямой к оси абсцисс, константа а равна величине ординаты lg i = 0 (т.е. при плотности тока i = 1). Величина перенапряжения водорода зависит от плотности тока, т.е. при данной силе тока, от истинных размеров поверхности катода. При одинаковой силе тока она больше на гладкой поверхности, чем на шероховатой. 3.4.4. Кислородная деполяризация Процессы коррозии с КИСЛОРОДНОЙ ДЕПОЛЯРИЗАЦИЕЙ могут протекать в том случае, если при данных условиях обратимый потенциал металла отрицательнее обратимого потенциала кислородного электрода (Me)обр < (О2)обр. (25) 54 Обратимый потенциал кислородного электрода может быть вычислен по уравнению Нернста: (О2)обр = (О2)ºобр + (RT2,3/4F)lg(pО2/a4ОH−) (26) где (О2)ºобр – стандартный обратимый потенциал кислородного электрода (при аOH– = 1 и рО2 = 1 ат); рО2 – парциальное давление кислорода; аОН– – активность гидроксильных ионов. Величина обратимого потенциала кислородного электрода (О2)ºобр при 25 oC для различных значений рН и рО2: рН среды ро2 , ат 0,21 1 0 + 1,218 + 1,229 7 + 0,805 + 0,815 14 + 0,391 + 0,400 Коррозия с кислородной деполяризацией является термодинамически более возможным процессом, чем коррозия с водородной деполяризацией, так как равновесный потенциал восстановления кислорода более положителен, чем равновесный потенциал выделения водорода. Коррозия металлов с кислородной деполяризацией является самым распространенным коррозионным процессом. Коррозии с кислородной деполяризацией подвергаются металлическая обшивка нефтезаводской аппаратуры и оборудования, детали аппаратов, соприкасающиеся с водой и нейтральными водными растворами солей (например, трубы конденсационно−холодильного оборудования), подземные трубопроводы, днища резервуаров и др. При коррозии металлов с кислородной деполяризацией катодный процесс определяется перенапряжением катодной реакции и скоростью подвода кислорода к электроду. Замедленность катодной реакции по схеме: О2 + 2Н2О + 4е 4ОН– (в щелочных или нейтральных растворах), или по схеме: О2 + 4Н+ + 4е 2Н2О (в слабо−кислых растворах) называется ПЕРЕНАПРЯЖЕНИЕМ ИОНИЗАЦИИ КИСЛОРОДА. При больших плотностях тока (при iк > 10–2 A/m2) dhe një shkallë të konsiderueshme të furnizimit me oksigjen në katodë, mbitensioni i jonizimit të oksigjenit ka një varësi logaritmike nga densiteti i rrymës 55 δ" = а"+ b" log i, (27) ku δ" është mbitensioni i jonizimit të oksigjenit; a" është një konstante që varet nga materiali i katodës dhe gjendja e sipërfaqes së saj, temperatura dhe faktorë të tjerë. Është përcaktuar numerikisht si vlera e mbitensionit në i = 1; b" është një konstante që nuk varet nga materiali katodë; përcaktohet nga barazia b" = 2RT/nF ∙ 2.3; në një temperaturë prej 20° dhe n = 1, b" = 0.117. Mekanizmi i reduktimit katodik të oksigjenit është mjaft kompleks dhe ndodh në disa faza: shpërbërja e oksigjenit në elektrolit, transferimi i oksigjenit të tretur në seksionet katodë të metalit, jonizimi i oksigjenit. Në ndryshim nga depolarizimi i hidrogjenit, për të cilin polarizimi i përqendrimit është praktikisht i parëndësishëm, në korrozionin e metaleve me depolarizimin e oksigjenit luan një rol shumë të rëndësishëm për shkak të shpejtësisë së kufizuar të furnizimit me oksigjen në katodë. Kjo shpjegohet nga tretshmëria e ulët e oksigjenit në tretësirën e elektrolitit dhe, rrjedhimisht, përqendrimi i ulët i tij, vështirësia e difuzionit të oksigjenit përmes një shtrese të palëvizshme të lëngut ngjitur me katodën. Me trazim të fortë të tretësirës, qarkullim intensiv ose në kushte që sigurojnë ajrim të konsiderueshëm të elektrolitit, procesi i korrozionit pengohet nga një mbitension i jonizimit të oksigjenit. Proceset e difuzionit ndikojnë në shkallën e korrozionit kur metali zhytet në një zgjidhje të qetë ose pak të trazuar. Kurba e përgjithshme e polarizimit katodik ka një formë komplekse (Fig. 12) dhe është totali i tre kurbave që karakterizojnë polarizimin gjatë jonizimit të oksigjenit (AB), polarizimit të përqendrimit (DE) dhe shkarkimit të joneve të hidrogjenit (GFH). Në pjesën e parë, shpejtësia e procesit katodik, d.m.th. sasia e oksigjenit e reduktuar për njësi të kohës është më e vogël se shpejtësia maksimale e mundshme e shpërndarjes së oksigjenit në sipërfaqe me anë të difuzionit. Në këtë drejtim, në këtë fushë shpejtësia e procesit kufizohet kryesisht nga ngadalësia e vetë procesit të reduktimit të oksigjenit. 56 − H F K E G (H2)arr 0 (O2)arr A D B C iд i Fig. 12 Kurba e polarizimit të katodës për procesin e depolarizimit të oksigjenit Me një rritje të mëtejshme të densitetit të rrymës, potenciali zhvendoset në drejtim negativ, së pari gradualisht, dhe pastaj befas (seksioni CDE). Një zhvendosje e mprehtë e potencialit në drejtim negativ shoqërohet me polarizimin e përqendrimit për shkak të një ulje të përqendrimit të oksigjenit. Me një vlerë të caktuar potenciale, bëhet i mundur një proces i ri katodik, i shoqëruar zakonisht me depolarizimin e hidrogjenit (seksioni EK). 3.5. Kontrollimi i faktorit të korrozionit Procesi i korrozionit elektrokimik të metaleve dhe lidhjeve përbëhet nga procese ose faza elementare. Shpejtësia aktuale e procesit të korrozionit varet drejtpërdrejt nga frenimi total i tij në çdo fazë. Përqindja e frenimit të procesit të përgjithshëm në secilën nga fazat e tij karakterizon SHKALLËN E KONTROLLIT TË PROCESIT në një fazë të caktuar. 57 quhet faza që përcakton shkallën e korrozionit, pasi ka rezistencën më të madhe në krahasim me fazat e tjera. PROCESI KONTROLLUES o2)arr imax I imax b b b a (o2)arr në A A imax (Me)arr (Me)arr K ∆K K K K I I S (о2)arr i I g g d Fig. 13. Diagramet e korrozionit të polarizimit për rastet kryesore praktike të monitorimit të proceseve të korrozionit elektrokimik 58 I në Në kushte praktike, ekzistojnë disa raste bazë të monitorimit të korrozionit elektrokimik të metaleve dhe lidhjeve, të vërtetuara nga N.D. Tomashov. Bekkerev I.V. "Metalet dhe lidhjet - Klasat, përbërja kimike", Pjesa 2, Ulyanovsk: Universiteti Teknik Shtetëror Ulyanovsk, 2007. - f. 630, http://www.bibliotekar.ru/spravochnik-73/index.htm. 3]. Semenova I.V. Mbrojtja nga korrozioni dhe korrozioni, M.: Fizmatlit, 2002, 335 f. 4]. Khaldeev G.V., Borisova T.F. "Përshkueshmëria nga hidrogjeni i metaleve dhe lidhjeve në proceset korrozioni-elektrokimike. Elektrokimi" - Vëllimi 30, Moskë, 1989 f. 232 5]. T. L. Lukanina, T. T. Ovchinnikova, V. Ya Sigaev, "Kimia e Përgjithshme" Pjesa II. Tutorial. Botimi i dytë, i korrigjuar dhe zgjeruar, 2006)