Хімічний опір будівельних матеріалів залежно від їх складу та будови. Технологічні характеристики матеріалів. Хімічний опір матеріалів лихачів владислав Олександрович до х Основний хід роботи
Міністерство загальної та професійної освіти
Російської Федерації
МОСКІВСЬКИЙ ДЕРЖАВНИЙ УНІВЕРСИТЕТ ІНЖЕНЕРНОЇ ЕКОЛОГІЇ
А.Г.Паршин В.С.Пахомов Д.Л.Лебедєв
Хімічний опір матеріалів та захист від корозії
Лабораторний практикум
За редакцією д-ра техн. наук О.О.Шевченка
Москва-1998
ББК35.11 Χ 46
Рецензенти:
кафедра корозії Московської державної академії нафти та газу ім. Губкіна;
канд. техн. наук А.С.Абрамов, екологічна фірма "Шанеко", Москва.
Допущено як навчальний посібник редакційно-видавничої ради Московського державного університету інженерної екології.
Паршин А.Г., Пахомов В.С, Лебедєв Д.Л.
Χ 46 Хімічний опір матеріалів та захист від корозії: Лабораторний практикум / За ред. А.А.Шевченко - М.: МДУІЕ, 1998.-80 с.; іл.8.
ISBN 5-230-11142-9
Практикум містить чотири лабораторні роботи з електрохімічної корозії та захисту металів: "Вплив структурної неоднорідності на корозію металів при відновленні іонів водню", "Корозія металів з кисневою деполяризацією", "Електродні потенціали", "Контактна корозія та катодна захист металів". Викладено основні уявлення теорії електрохімічної корозії металів та методику проведення корозійних досліджень. Призначений для студентів 3 та 4 курсів денних факультетів, які вивчають курс хімічного опору матеріалів та захисту від корозії.
ISBN 5-230-11142-9 УДК620.193 ББК 35.11
© А.Г.Паршин, В.С.Пахомов, Д.Л.Лебедєв.1998
© МДУІЕ, 1998
Передмова
Лабораторний практикум написаний відповідно до програми курсу "Хімічний опір матеріалів та захист від корозії", передбаченого навчальним планом низки спеціальностей. До практикуму увійшли роботи з електрохімічної корозії металів, які розроблені на кафедрі "Композиційні матеріали та захист від корозії".
Кожна лабораторна робота розпочинається з теоретичної частини. Розглянуто механізм, термодинаміка та кінетика корозії при катодному відновленні іонів водню, кисневої деполяризації, рівноважні та нерівноважні процеси на межі розділу метал-електроліт, теорія електродних потенціалів.
При викладі теоретичних питань використано сучасні електрохімічні уявлення про механізм та кінетику корозійних процесів.
Виконання лабораторних робіт дозволяє студентам глибше засвоїти основи вчення про корозію та захист металів, а також прищеплює навички проведення елементарних лабораторних корозійно-електрохімічних досліджень.
Ведення лабораторного журналу
1) назва та мета роботи;
2) схему встановлення;
3) таблицю з результатами дослідів та їх обробки (розрахунки, графіки);
4) висновки.
Вести журнал слід акуратно і одночасно начисто, тобто. так, як ведуть записи всіх науково-дослідних робіт та випробувань.
Схема установки має бути наочна і зрозуміла без усних пояснень.
Необхідно навести умови досвіду: випробувані матеріали, площа поверхні зразків, склад та концентрація електролітів, температура та ін.
Результати випробувань слід записувати в наперед складені таблиці, форми яких наведені в описі робіт.
Під час запису різних величин необхідно вказувати їх розмірність. Результати вимірювань, як правило, підлягають додатковій обробці – аналітичній та графічній (обчислення масового, об'ємного та глибинного показників корозії, потенціалів та ін.). У разі необхідно навести розрахункову формулу і один розрахунок повністю, тобто. з підстановкою у формулу дослідних величин, а інших аналогічних розрахунків - лише кінцеві результати.
На підставі отриманих та відповідним чином опрацьованих результатів слід зробити короткі висновки про виконану роботу. Після закінчення журнал з експериментальними даними подати на візу викладачеві.
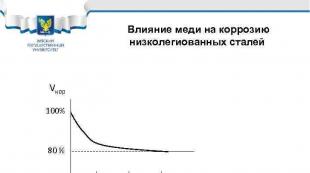
 Вплив міді на корозію низьколегованих сталей Vкор 100% 80 % 0, 1 0, 2 0, 3 % Cu Приклади сталей: 10 ХСНД, 10 Г 2 С 1 Д, 10 ХДНП, 09 Г 2 Д, 18 Г 2 АФ(Д)
Вплив міді на корозію низьколегованих сталей Vкор 100% 80 % 0, 1 0, 2 0, 3 % Cu Приклади сталей: 10 ХСНД, 10 Г 2 С 1 Д, 10 ХДНП, 09 Г 2 Д, 18 Г 2 АФ(Д)
 Класифікація корозійностійких сталей 1. Корозійностійкими (нержавіючими) сталями та сплавами називаються матеріали, що опираються електрохімічній корозії в електролітах. 2. Основним легуючим елементом корозійностійкого легування є хром. 3. Хром у нержавіючі сталі вводиться відповідно до правила Таммана. 4. Залежно від середовищ, у яких ці сталі застосовуються, розрізняють п'ять груп корозійностійких (нержавіючих) сталей та сплавів.
Класифікація корозійностійких сталей 1. Корозійностійкими (нержавіючими) сталями та сплавами називаються матеріали, що опираються електрохімічній корозії в електролітах. 2. Основним легуючим елементом корозійностійкого легування є хром. 3. Хром у нержавіючі сталі вводиться відповідно до правила Таммана. 4. Залежно від середовищ, у яких ці сталі застосовуються, розрізняють п'ять груп корозійностійких (нержавіючих) сталей та сплавів.
 Сталі першої групи можуть працювати тільки в умовах закритої атмосфери і при підводній корозії при обов'язковому періодичному висушуванні. В умовах відкритої атмосфери та постійної підводної корозії (особливо в гарячій воді), а також підземної корозії ці сталі піддаються піттинговій корозії. До таких сталей відносяться хромисті сталі: 08 Х 13, 09 Х 13, 08 Х 17 Г (феритні), 10 Х 13, 12 Х 13 (мартенситно-феритні), 20 Х 13, 30 Х 13, 40 Х 13 (мартенситні) . А також хром-марганцеві та хром-нікелеві сталі з економним легуванням по нікелю (2 -4%) 15 Х 17 АГ 14, 10 Х 14 АГ 15, 10 Х 14 Г 14 Н 3 Т, 12 Х 17 Г 14 Н 3 , 08 Х 18 Г 8 Н 2 Т
Сталі першої групи можуть працювати тільки в умовах закритої атмосфери і при підводній корозії при обов'язковому періодичному висушуванні. В умовах відкритої атмосфери та постійної підводної корозії (особливо в гарячій воді), а також підземної корозії ці сталі піддаються піттинговій корозії. До таких сталей відносяться хромисті сталі: 08 Х 13, 09 Х 13, 08 Х 17 Г (феритні), 10 Х 13, 12 Х 13 (мартенситно-феритні), 20 Х 13, 30 Х 13, 40 Х 13 (мартенситні) . А також хром-марганцеві та хром-нікелеві сталі з економним легуванням по нікелю (2 -4%) 15 Х 17 АГ 14, 10 Х 14 АГ 15, 10 Х 14 Г 14 Н 3 Т, 12 Х 17 Г 14 Н 3 , 08 Х 18 Г 8 Н 2 Т
 Друга група корозійностійких (нержавіючих) сталей застосовується в сольових середовищах при невисоких температурах, зокрема при морській корозії. Підвищена корозійна стійкість досягається додатковим економним легуванням сталей Ni (5 – 8%). Приклади: 09 Х 15 Н 8 Ю, 07 Х 16 Н 6, 08 Х 17 Н 5 М 3 (сталь використовується в сірчанокислих середовищах), 09 Х 17 Н 7 Ю 1 (сталі застосовуються в умовах морської корозії).
Друга група корозійностійких (нержавіючих) сталей застосовується в сольових середовищах при невисоких температурах, зокрема при морській корозії. Підвищена корозійна стійкість досягається додатковим економним легуванням сталей Ni (5 – 8%). Приклади: 09 Х 15 Н 8 Ю, 07 Х 16 Н 6, 08 Х 17 Н 5 М 3 (сталь використовується в сірчанокислих середовищах), 09 Х 17 Н 7 Ю 1 (сталі застосовуються в умовах морської корозії).
 Стали для застосування в середовищі середньої корозійної агресивності Під середовищами із середньою корозійною агресивністю розуміють розчини солей за різних температур, а також слабкі розчини деяких кислот. Стали третьої групи – найпоширеніші нержавіючі сталі широкого застосування. Серед цих сталей можна виділити: а) сталі – замінники високонікелевих: 15 Х 25 Т, 15 Х 28, 08 Х 22 Н 6 Т, 12 Х 21 Н 5 Т. б) стали з оптимальним співвідношення хрому до нікелю (Cr: Ni = 18: 9, 18: 10): 12 Х 18 Н 9 Т і 12 Х 18 Н 10 Т, 17 Х 18 Н 9, 12 Х 18 Н 10 Б, 08 Х 18 Н 10, 12 Х 18 Н 12 Т, 08 Х 18 Н 12 Б, 06 Х 18 Н 11 і т.д.
Стали для застосування в середовищі середньої корозійної агресивності Під середовищами із середньою корозійною агресивністю розуміють розчини солей за різних температур, а також слабкі розчини деяких кислот. Стали третьої групи – найпоширеніші нержавіючі сталі широкого застосування. Серед цих сталей можна виділити: а) сталі – замінники високонікелевих: 15 Х 25 Т, 15 Х 28, 08 Х 22 Н 6 Т, 12 Х 21 Н 5 Т. б) стали з оптимальним співвідношення хрому до нікелю (Cr: Ni = 18: 9, 18: 10): 12 Х 18 Н 9 Т і 12 Х 18 Н 10 Т, 17 Х 18 Н 9, 12 Х 18 Н 10 Б, 08 Х 18 Н 10, 12 Х 18 Н 12 Т, 08 Х 18 Н 12 Б, 06 Х 18 Н 11 і т.д.
 Стали для застосування в середовищах з підвищеною корозійною агресивністю Такі стали розроблялися з метою підвищення хімічного опору в гарячих розчинах Na. Cl та в розчинах кислот. Для підвищення стійкості сталей застосовується додаткове легування їх молібденом та міддю, причому у сталях цієї групи часто прагнуть зберегти аустенітну структуру, зручну в технологічному відношенні, що потребує додаткового легування сталей нікелем. У зв'язку з високим вмістом легуючих компонентів, насамперед нікелю, стали цієї групи досить дорогими. Прикладом сталей групи є: 10 Х 17 Н 13 М 2 Т 08 Х 17 Н 13 М 3 Т, 08 Х 17 Н 15 М 3 Т, 04 Х 28 МДТ, 03 Х 28 МДТ, 06 Х 28 МТ.
Стали для застосування в середовищах з підвищеною корозійною агресивністю Такі стали розроблялися з метою підвищення хімічного опору в гарячих розчинах Na. Cl та в розчинах кислот. Для підвищення стійкості сталей застосовується додаткове легування їх молібденом та міддю, причому у сталях цієї групи часто прагнуть зберегти аустенітну структуру, зручну в технологічному відношенні, що потребує додаткового легування сталей нікелем. У зв'язку з високим вмістом легуючих компонентів, насамперед нікелю, стали цієї групи досить дорогими. Прикладом сталей групи є: 10 Х 17 Н 13 М 2 Т 08 Х 17 Н 13 М 3 Т, 08 Х 17 Н 15 М 3 Т, 04 Х 28 МДТ, 03 Х 28 МДТ, 06 Х 28 МТ.
 Сплави на нікелевій основі для дуже агресивних середовищ Під середовищами з дуже високою агресивністю розуміються гарячі розчини сірчаної та соляної кислот. У таких агресивних середовищах із металевих матеріалів найбільш стійкими є сплави на основі нікелю. Наприклад, сплав ХН 65 МВ стійкий при підвищеній температурі в сірчанокислих і солянокислих середовищах, концентрованої оцтової кислоти. Сплав Н 70 МФ рекомендований для використання в сірчанокислих, солянокислих розчинах, сплав більш стійкий до міжкристалітної корозії.
Сплави на нікелевій основі для дуже агресивних середовищ Під середовищами з дуже високою агресивністю розуміються гарячі розчини сірчаної та соляної кислот. У таких агресивних середовищах із металевих матеріалів найбільш стійкими є сплави на основі нікелю. Наприклад, сплав ХН 65 МВ стійкий при підвищеній температурі в сірчанокислих і солянокислих середовищах, концентрованої оцтової кислоти. Сплав Н 70 МФ рекомендований для використання в сірчанокислих, солянокислих розчинах, сплав більш стійкий до міжкристалітної корозії.
 Підвищення густини бетону 4. Введення полімерних добавок 4. 1. введення невеликої кількості 0, 2 - 3% полімерних добавок у бетонну суміш (латекси, полімерні смоли); 4. 2. виготовлення бетонів на основ полімерного в'яжучого (полімеррозчини та полімербетони); Поставляється у вигляді сухої суміші та затверджувача в банках. 4. 3. просочування готових бетонів та залізобетонних виробів полімерними складами або мономерами з подальшою полімеризацією їх безпосередньо в тілі бетону (бетонополімери); 4. 4. армування бетону полімерними волокнами (отримання фібробетонів)
Підвищення густини бетону 4. Введення полімерних добавок 4. 1. введення невеликої кількості 0, 2 - 3% полімерних добавок у бетонну суміш (латекси, полімерні смоли); 4. 2. виготовлення бетонів на основ полімерного в'яжучого (полімеррозчини та полімербетони); Поставляється у вигляді сухої суміші та затверджувача в банках. 4. 3. просочування готових бетонів та залізобетонних виробів полімерними складами або мономерами з подальшою полімеризацією їх безпосередньо в тілі бетону (бетонополімери); 4. 4. армування бетону полімерними волокнами (отримання фібробетонів)

 Модуль 7. Методи захисту металів від електрохімічної корозії. Лекція 7. 3 ЕЛЕКТРОХІМІЧНИЙ ЗАХИСТ МЕТАЛІВ ВІД КОРОЗІЇ Катодний захист полягає у зміщенні потенціалу металу кородируючою конструкції в негативну сторону за рахунок приєднання його до негативного полюса джерела струму.
Модуль 7. Методи захисту металів від електрохімічної корозії. Лекція 7. 3 ЕЛЕКТРОХІМІЧНИЙ ЗАХИСТ МЕТАЛІВ ВІД КОРОЗІЇ Катодний захист полягає у зміщенні потенціалу металу кородируючою конструкції в негативну сторону за рахунок приєднання його до негативного полюса джерела струму.
 Модуль 7. Методи захисту металів від електрохімічної корозії. Лекція 7. 3 Корозійна діаграма катодного захисту
Модуль 7. Методи захисту металів від електрохімічної корозії. Лекція 7. 3 Корозійна діаграма катодного захисту
 Модуль 7. Методи захисту металів від електрохімічної корозії. Лекція 7. 2 Протекторний захист ґрунтується на особливостях корозії двох металів у контакті. Згідно з теорією контактної корозії, при контакті позитивного металу М 2 з більш негативним М 1 потенціал металу М 2 зміщується в негативну сторону, корозія його при цьому зменшується або повністю припиняється.
Модуль 7. Методи захисту металів від електрохімічної корозії. Лекція 7. 2 Протекторний захист ґрунтується на особливостях корозії двох металів у контакті. Згідно з теорією контактної корозії, при контакті позитивного металу М 2 з більш негативним М 1 потенціал металу М 2 зміщується в негативну сторону, корозія його при цьому зменшується або повністю припиняється.
 Модуль 7. Методи захисту металів від електрохімічної корозії. Лекція 7. 3 Анодний захист застосовується лише для металів, схильних до пасивації у корозійному середовищі. Вона зводиться до зміщення потенціалу металу з області активного розчинення область пасивації за допомогою зовнішнього джерела струму.
Модуль 7. Методи захисту металів від електрохімічної корозії. Лекція 7. 3 Анодний захист застосовується лише для металів, схильних до пасивації у корозійному середовищі. Вона зводиться до зміщення потенціалу металу з області активного розчинення область пасивації за допомогою зовнішнього джерела струму.
 Модуль 7. Методи захисту металів від електрохімічної корозії. Лекція 7. 3 Корозійна діаграма анодного захисту
Модуль 7. Методи захисту металів від електрохімічної корозії. Лекція 7. 3 Корозійна діаграма анодного захисту
МІНІСТЕРСТВО ОСВІТИ І НАУКИ РОСІЙСЬКОЇ ФЕДЕРАЦІЇ ФЕДЕРАЛЬНА ДЕРЖАВНА ОСВІТА ЗАСТАВА ВИЩОЇ ПРОФЕСІЙНОЇ ОСВІТИ «САНКТ-ПЕТ НІВЕРСИТЕТ РОСЛИННИХ ПОЛІМЕРІВ» Т. Л. Луканіна, І. С. Михайлова, М. А. Радін ХІМІЧНИЙ ОПОР МАТЕРІАЛІВ І ЗАХИСТ ВІД КОРОЗІЇ −Петербург 2014 1 УДК 546(075) ББК 24.1я7 Л 840 Луканіна Т. Л., Михайлова І. С., Радін М. А. Хімічний опір матеріалів та захист від корозії: навч. посібник. - СПб.: СПбДТУРП, 2014. - 85 с. У посібнику дано сучасні уявлення про термодинаміку та кінетику хімічної корозії, механізми та різні види електрохімічної корозії з урахуванням середовищ та умов, характерних для обладнання хімічної промисловості. Викладено наукові принципи створення корозійностійких сталей та сплавів, способи захисту від корозії конструкційних матеріалів. Навчальний посібник пропонується для студентів технічних вузів та факультетів, які навчаються за технологічними спеціальностями. Може бути корисним для аспірантів, наукових та інженерно-технічних працівників хімічних підприємств, проектних та дослідницьких організацій, які займаються захистом металів від корозії. Рецензенти: д-р хім. наук, проф. кафедри СПбГІТМО хім. факультету Слободів А. А.; канд. хім. наук, доц. кафедри органічної хімії СПбДТУРП Євдокимов А. Н. Рекомендовано до видання Редакційно-видавничою радою університету як навчальний посібник. Редактор та коректор Т. А. Смирнова Техн. Редактор Л.Я.Тітова Темплан 2014, поз. 45 _____________________________________________________________ Підписано до друку 14.04.14 Формат 60×84/16. Папір тип №1. Друк офсетний. Печ.л.5,5. Уч.-изд.л.5,5.Тіраж 100 екз.Изд № 45. Ціна «С».Заказ Різограф Санкт-Петербурзького державного технологічного університету рослинних полімерів, СПб., 198095, вул. Івана Чорних, 4 Санкт-Петербурзький державний технологічний університет рослинних полімерів, 2014 Луканіна Т. Л., Михайлова І. С., Радін М. А., 2014 2 ВСТУП КОРОЗІЇ матеріалів називається мимовільне руйнування внаслідок хімічної або електрохімічної взаємодії з навколишнім середовищем. Руйнування металів і сплавів від корозії відрізняється від інших видів мимовільного руйнування, наприклад, від ерозії або зношування, які викликаються механічною взаємодією з тілом чи середовищем. У більшості металів і сплавів в умовах їх експлуатації стійкішим є окислений (іонний) стан, в який вони переходять в результаті корозії. Термін "корозія" походить від латинського слова "corrodere", що означає "роз'їдати". Втрати від корозії прийнято ділити на прямі та непрямі. До ПРЯМИХ Втрат відносять: − вартість металу, який перетворюється на продукти корозії, а також вартість обладнання, що прокородує, що вийшло з ладу до вичерпання передбаченого технічними умовами амортизаційного терміну. При цьому, як правило, вартість виготовлення обладнання набагато перевищує вартість металу, з якого його виготовлено; − збільшення припусків на корозію, що призводить не тільки до збільшення витрати металу, а й до погіршення конструкції. Наприклад, для трубопроводу при тому самому зовнішньому діаметрі підвищений припуск металу тягне зменшення корисного перерізу, і, отже, зниження пропускної здатності труби; − витрати на захисні заходи у вигляді металевих та неметалічних покриттів, корозійно-стійких матеріалів, інгібіторів та ін. − недовиробництвом продукції внаслідок зупинок на ремонт зруйнованого корозією обладнання; − можливістю аварій, вибухів та пожеж; − витоком продукції при утворенні внаслідок корозії наскрізних отворів у стінках обладнання; 3 − зменшенням перерізу трубопроводів внаслідок відкладення продуктів корозії, що потребує додаткової витрати енергії для підтримки необхідної потужності потоку рідини; − зниженням якості продукції внаслідок забруднення продуктами корозії. Наука про корозію та захист металів вивчає взаємодію металів та сплавів з корозійним середовищем, встановлює механізм цієї взаємодії та її загальні закономірності. Науковою основою для вчення про корозію та захист металів є металознавство та фізична хімія. Своєю кінцевою практичною метою ця наука має захист від корозійного руйнування металів під час їх обробки та експлуатації обладнання. Глава 1. ЗАГАЛЬНІ ВІДОМОСТІ ПРО КОРОЗІЮ 1.1. Термодинаміка та кінетика корозійних процесів Першопричиною корозії є термодинамічна нестійкість металів у різних середовищах за даних зовнішніх умов. Принципова можливість мимовільного перебігу процесу корозії визначається законами термодинаміки. Перебіг будь-якого мимовільного процесу супроводжується зменшенням вільної енергії. Оцінити можливість мимовільного перебігу процесів корозії можна змінити ізобарно-ізотермічного потенціалу ΔG1. Мимовільний процес окислення металу – корозія – можливий, якщо ∆G< 0, то есть знак ―минус‖ означает, что изобарно−изотермический потенциал убывает и свободная энергия системы уменьшается. Наоборот, если ∆G > 0, то знак "плюс" у ізобарно-ізотермічного потенціалу відповідає збільшенню вільної енергії, що вказує на неможливість мимовільного перебігу цієї реакції. ІЗОБАРНО-ІЗОТЕРМІЧНИЙ ПОТЕНЦІАЛ (ΔG) – енергія Гіббса, один із термодинамічних потенціалів, рівний ΔG = ΔН - TΔS, де ΔН, ΔS, Т - ентальпія, ентропія, термодинамічна температура системи. ΔG пропорційний числу частинок у системі. Віднесений до однієї частки, він називається хімічним потенціалом. ΔG зручний для опису процесів, в яких можливий обмін між речовиною та оточуючими тілами. В ізотермічно рівноважному процесі, що відбувається при постійному тиску, це повна робота системи за вирахуванням роботи проти сил зовнішнього тиску. Вона дорівнює спаду ізобарно-ізотермічного потенціалу (тобто максимально корисна робота). 1 4 Однак, хоча термодинаміка дає можливість визначити, наскільки система, що вивчається, віддалена від стану рівноваги і здатна до мимовільної реакції, це не свідчить про швидкість даного процесу. Швидкість корозії визначається його кінетикою. Особливістю корозійних процесів є їхній гетерогенний характер. Це з тим, що руйнація металу відбувається межі розділу двох фаз, мають різний агрегатний стан, наприклад межі метал – рідина, метал – газ. У таких умовах визначальним фактором для кінетики хімічної взаємодії є або швидкість протікання хімічної взаємодії, або підведення реагентів з обсягів дотичних фаз на межу розділу, де відбувається корозійна взаємодія, та відведення продуктів реакції у зворотному напрямку. У першому випадку швидкість корозії визначається кінетичним контролем, у другому дифузійним. У деяких випадках швидкість корозії визначається змішаним дифузійно-кінетичним контролем. Для процесу процесу швидкість хімічної реакції і швидкість дифузії рівні. Корозійний процес складається з кількох стадій, які можуть протікати або послідовно або паралельно. Сумарна швидкість корозійного процесу, що складається з декількох послідовних стадій, визначається швидкістю найповільнішої стадії. При паралельному перебігу окремих стадій процесу сумарна швидкість корозії визначається найшвидшою стадією. Тому у випадку, якщо одна із стадій протікає значно швидше за іншу, другою стадією можна знехтувати. 1.2. Класифікація процесів корозії за механізмом протікання Незалежно від величезної різноманітності умов протікання корозійних процесів вони можуть бути класифіковані за двома основними ознаками: – за механізмом перебігу процесу – за характером корозійного руйнування. Розрізняють ХІМІЧНУ та ЕЛЕКТРОХІМІЧНУ корозію. 5 має місце в сухих газах (газова корозія) і рідких неелектролітах2 (корозія в неелектролітах). Якщо середовище не є електролітом (сухі гази або рідкі неелектроліти), то обмін електронами здійснюється безпосередньо між металом-відновником (red) і будь-якою речовиною, що містить електронегативний елемент, здатним відтягнути на себе електрони - окислювачем (ox), наприклад: Fe + Сl2 → FeCl2 (1) ХІМІЧНА КОРОЗІЯ red Баланс передачі електронів ox Feº − 2ē → Fe2+ 1 Сl2º + 2ē → 2Cl− Feº + 2Сlº = Fe2+ + 2Cl− ЕЛЕКТРОХІМІЧНА КОРОЗІЯ – це процес взаємодії іонізація атомів металу та відновлення окисного компонента корозійного середовища протікають не в одному акті та їх швидкості залежать від електродного потенціалу. Відмінними рисами електрохімічного процесу корозії є такі: – одночасне перебіг двох роздільних процесів – окислювального (розчинення металу) та відновного (виділення водню, відновлення кисню, виділення металу з розчину та ін.); – процес розчинення металу супроводжується спрямованим переміщенням електронів у металі та іонів в електроліті, тобто. виникнення електричного струму; – продукти корозії утворюються внаслідок вторинних реакцій. Окисно-відновні процеси, що протікають при електрохімічній корозії, можуть бути представлені у вигляді наступних реакцій: Ме - n → → (+) (2) n - R (ox) + n → R (red) (катодний процес), (3 ) де R(ox) - окислювач; НЕЕЛЕКТРОЛІТИ - речовини, водні розчини та розплави яких не проводять електричний струм. Вони містять ковалентні неполярні чи малополярні зв'язки, які розпадаються на іони. Електричний струм не проводять гази, тверді речовини (неметали), органічні сполуки (цукроза, бензин, спирт). 2 6 R n−(red) – відновлена форма окислювача; nē – кількість переданих електронів. Як приклад електрохімічної корозії можна навести процес окислення (іржавіння) заліза під впливом води: Fe − 2ē → Fе2+ (анодний процес – розчинення заліза) H2O + ½О2 +2ē → 2OH– (катодний процес – відновлення кисню) −−−−−− −−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−Fe 2+ + 2OH – → Fe(OH)2 (утворення продуктів корозії) Реакції (2) і (3) протікають сполучено, але підпорядковуються своїм кінетичним закономірностям. У цьому необхідно дотримання умов стаціонарності процесу, тобто. рівності швидкостей окислення металу та відновлення окислювача. Дані реакції можуть бути територіально розділені – протікати різних ділянках поверхні. З умов стаціонарності випливає, що досить загальмувати одну із сполучених реакцій, щоб швидкість всього процесу зменшилася. 1.3. Класифікація процесів корозії за характером корозійного руйнування За характером корозійного руйнування металу розрізняють ЗАГАЛЬНУ І МІСЦЕВУ КОРОЗІЮ. Загальна корозія поширюється по всій поверхні металу, вона може бути рівномірною та нерівномірною (рис. 1. - 1,2). 1 2 4 5 7 8 3 6 9 Мал. 1. Види корозійних руйнувань До загальної відносяться багато випадків корозії, наприклад, коли внаслідок впливу на метал електролітів продукти корозії не залишаються на поверхні металу. Так, інтенсивна загальна корозія спостерігається при взаємодії міді з азотною кислотою, 7 заліза з соляною кислотою, алюмінію з їдкими лугами, цинку з сірчаною кислотою та ін. Рівномірна загальна корозія є одним з найменш небезпечних видів корозії за умови, що швидкість розчинення корозійних руйнувань вбирається у норм, визначених вимогами до експлуатації апарату, приладу тощо. При достатній товщині металу корозія мало позначається на зменшенні механічної міцності конструкції при рівномірно розподілених напругах перерізу (розтягування, стиснення). Однак загальна корозія може бути небезпечною при роботі деталей на вигин та кручення, так як у цьому випадку руйнуються найбільш навантажені шари. МІСЦОВА КОРОЗІЯ − викликає руйнування лише деяких ділянок поверхні металу, а решта не піддається руйнуванню або швидкість руйнування дуже мала. Прояви місцевої корозії дуже різноманітні і можуть мати різний ступінь нерівномірності, у зв'язку з чим місцева корозія ділиться на корозію плямами, виразками, крапками (ПІТТИНГ), виборчу корозію, корозію, корозійно. Міжкристалічна корозія плямами, виразками, точками - види місцевої корозії різняться за співвідношенням діаметра зруйнованої ділянки до її глибини (рис.1., 4-6). При корозії плямами діаметр зруйнованої ділянки набагато більший за його глибину, при виразковому руйнуванні величина діаметра зруйнованої ділянки порівнянна з його глибиною, а при точковій корозії діаметр зруйнованої ділянки набагато менше його глибини. У деяких випадках при точковій корозії можливе наскрізне руйнування, що є особливо небезпечним для трубопроводів, резервуарів і т.д. Точкова корозія типова для металів, здатних до пасивації – гальмування (хром, алюміній, нержавіючі сталі та ін.). Пасивний стан металів може порушуватися в окремих більш ослаблених ділянках поверхні, наприклад, у порах захисної плівки, в місцях інтерметалічних включень (оксиди, шлаки та ін.). ВИБОРЧА КОРОЗІЯ – підрозділяється на: компонентно-виборчу (селективну) або структурно-виборчу (інтеркристалітну). 8 Прикладом КОМПОНЕНТНО-ВИБОРЧОЇ КОРОЗІЇ може бути знецинкування латунів. Знецинкування латуней3 полягає в тому, що в електроліт (зазвичай нейтрального або слабокислого значення рН) цинк переходить інтенсивніше, ніж мідь. На поверхні латуні утворюється пухкий шар міді, що сприяє посиленню електрохімічної корозії (виникнення мікрогальванічного елемента). Прикладом СТРУКТУРНО-ВИБОРЧОЇ корозії є корозія сірих чавунів. Структурно-виборча корозія сірих чавунів полягає у переважному руйнуванні феритної складової, унаслідок чого утворюється скелет із графіту, заповнений продуктами корозії (рис.1-3). При цьому механічна міцність чавунів різко знижується. Міжкристалічна корозія характеризується вибірковим руйнуванням металу по межах зерен (рис. 1-7). Внаслідок цього при малій зміні маси та зовнішнього вигляду може відбуватися дуже значна втрата властивостей міцності металу (рис. 2). 40 Мал. 2. Втрата властивостей міцності металу при різних видах корозійного руйнування 1 - рівномірна корозія; 2 – міжкристалітна корозія 20 Втрата в, % 60 2 0 1 2 4 Втрата маси, кг/см2 У крайніх випадках при повному руйнуванні меж зерен внаслідок міжкристалітної корозії сплав може розсипатися на порошок. Корозійне розтріскування являє собою випадок місцевої корозії, при якому руйнування відбувається в напрямку, перпендикулярному найбільшій напругою, що розтягує (рис.1 - 8). 3 Латунь – мідно-цинковий сплав. 9 Характерно, що при цьому корозійна тріщина може поширюватися на межі зерен або перерізати тіло кристала. Руйнування від корозійної втоми (при одночасному впливі корозійного середовища та знакозмінних або циклічних навантажень) протікає за аналогічним типом, як і при корозійному розтріскуванні. Підповерхнева корозія, починаючись з поверхні, поширюється переважно під поверхнею, що часто призводить до розшаровування і спучування металу (рис.1-9), наприклад, утворення пухирів на неякісному листовому прокаті в процесі травлення або корозії при експлуатації судин і апаратів. 1.4. Показники корозії Для кількісного вираження швидкості корозії служать показники корозії: МАСОМЕТРИЧНИЙ, ОБСЯГНИЙ, ГЛУБИННИЙ, МЕХАНІЧНИЙ, СТРУМОВИЙ. 2 МАСОМЕТРИЧНИЙ показник корозії (Кm, г/м ∙год) являє собою зміну маси металу в результаті корозії, віднесену до одиниці поверхні (s) зразка та до одиниці часу. Зміна маси зразка визначається зазвичай як різницю між масою зразка до випробування (mо) та його масою після випробування (m1) протягом часу (τ) зі зняттям продуктів корозії (зменшення маси металу). K m = (m0 − m1)/s ∙ τ. (4) 3 2 ОБ'ЄМНИЙ показник корозії (Kν , cм /м год) являє собою обсяг (Vo) поглиненого або виділеного в процесі корозії газу, віднесений до одиниці поверхні металу та до одиниці часу K m = V0 /s ∙ τ . (5) ГЛУБИННИЙ показник корозії (П, мм/рік) враховує зменшення товщини металу внаслідок корозії, виражене в лінійних одиницях і віднесене до одиниці часу. Цей показник зручний при порівнянні корозії металів із різними щільностями. Перехід від масометричного показника до глибинного може бути зроблений у разі рівномірної корозії за формулою П = K m ∙ 8,76 /γ , (6) де γ – щільність металу. МЕХАНІЧНИЙ показник корозії визначається за зміною одного з основних показників механічних властивостей металу за певний час корозійного процесу і виражається у відносних одиницях або у відсотках. Якщо як механічний показник використовується межа міцності, то зміна показника міцності (Kσ) визначається за формулою Kσ = Δσb/σb, (7) де σb – межа міцності до корозії, ∆σb – зміна межі міцності металу після корозії протягом часу τ. Токовий показник корозії дозволяє, користуючись формулою Фарадея, підрахувати кількість металу, що прокородував, виходячи з величини струму корозії Km = IA /nF, (8) де Km – швидкість корозії зі зміни маси металу; I – сила струму; F – константа Фарадея; n – валентність металу; А – атомна маса металу. При якісній та кількісній оцінці корозійної стійкості металів рекомендується користуватися десятибальною шкалою4. Слід зазначити, що, хоча ця шкала оцінки корозійної стійкості металів і набула більшого поширення, особливо в хімічній промисловості, вона не є універсальною, оскільки для обладнання різного призначення допустимі швидкості корозії можуть відрізнятися на порядок і більше. Таблиця 1 Десятибальна шкала корозійної стійкості металів і сплавів ГРУПА СТІЙКОСТІ Повністю стійкі Стійкі Знижено-стійкі Малостійкі Нестійкі ШВИДКІСТЬ КОРОЗІЇ, ММ/РІК менше 0,005 до 0,00 до 0 01 до 0,05 понад 0, 05 до 0,1 понад 0,1 до 0,5 понад 0,5 до 1,0 понад 1,0 до 5,0 понад 5,0 до 10,0 понад 10,0 БАЛ 1 2 3 4 5 6 7 8 9 10 Єдина система захисту від корозії та старіння. Споруди підземні. Загальні вимоги щодо захисту від корозії. ГОСТ 9.602-2005, http://www.gosthelp.ru/gost/gost206.html 4 11 Глава 2. ХІМІЧНА КОРОЗІЯ МЕТАЛІВ Хімічна корозія металів є мимовільним процесом їх руйнування внаслідок хімічної взаємодії із зовнішнім середовищем. Відмінними рисами хімічного процесу корозії є такі: – здійснення процесів окислення та відновлення в одну стадію; - Відсутність електричного струму; - Утворення продуктів корозії безпосередньо на тій ділянці поверхні, де відбувається його руйнування. ХІМІЧНА КОРОЗІЯ є гетерогенною реакцією взаємодії рідкої (ж) або газової фази (г) з твердою фазою (т) – металом і ділиться на 2 види (газова корозія, корозія у рідких неелектролітах). 2.1. Газова корозія На практиці саме газова корозія має найбільше значення. Найчастіше вона проявляється як корозія металевих матеріалів при високій температурі (у десятки разів перевищує температуру кипіння води) в атмосфері кисневмісних газів. З цієї причини її називають високотемпературною корозією або високотемпературним окисленням. На поверхні виробів з металів і сплавів в результаті їх взаємодії з таким корозійним середовищем утворюється плівка продуктів, що є різними оксидами металів. У той же час в залежності від умов експлуатації можуть утворюватися й інші сполуки, наприклад, сірчисті метали5. Наприклад, у нафтопереробній промисловості газової корозії піддаються, наприклад, труби печей, стінки реакторів, реакційні колони та інша апаратура, що знаходяться під впливом сірководню, окису вуглецю, азоту, кисню та інших газів при високих температурах. Термін «газова корозія» відносять до руйнування металів, що викликається дією парів і газів при високих температурах, хоча його можна застосовувати і до більш низьких температур при вс5 СЕРНИСТІ МЕТАЛИ, те ж, що сульфіди. 12 ловії, що на металі не конденсується плівка рідини, що проводить електричний струм. На поверхні виробів з металів і сплавів в результаті їх взаємодії з корозійним середовищем утворюється плівка продуктів, що є різними сполуками металів. Загалом процес хімічної корозії можна описати, наприклад, реакцією окислення металу нагрітим (T >>373K) газом (Г2): 2Ме + nГ2 → 2Men+Гn. У середовищі кисню такий процес може бути описаний наступним рівнянням Ме(Т) + ½ О2 ↔ МеО(Т). Ця реакція за високих температур рівноважна. Зворотний процес називається ДИСОЦІАЦІЯ ОКСИДУ. Кисень, присутній у газовому середовищі, характеризується величиною парци6 ального тиску (рО2). Хімічна реакція окислення металу перебуватиме в рівновазі з реакцією дисоціації оксиду, якщо парціальний тиск кисню (рО2) і ПРУГІСТЬ ДИССОЦІАЦІЇ ОКСИДУ (рМеО)7 стануть рівні (рМеО = рО2) 1. При рО2 > рМе реа При рМеО > рО2 реакція протікає переважно у напрямку. Окислення металів - багатостадійний гетерогенний процес, завдяки якому спочатку на поверхні утворюється моно-, а потім полімолекулярний шар оксиду, так звана оксидна плівка. Зростання її товщини може відбуватися або в обох напрямках одночасно (металу та газового середовища), або переважно в одному (рис. 3). ПАРЦІАЛЬНИЙ ТИСК (від позднелат. partialis - частковий), тиск, який мав би газ, що входить до складу газової суміші, якби він один займав об'єм, що дорівнює об'єму суміші при тій же температурі. 7 Рівноважний парціальний тиск кисню для реакцій освіти або ТЕРМІЧНОЇ ДИССОЦІАЦІЇ ОКСИДІВ називається ПРУГОСТЮ ДИССОЦІАЦІЇ ОКСИДУ. Її величина пов'язана з константою рівноваги реакції і, отже, є функцією температури. 6 13 а б Рис. 3. Схема іонно-електронного механізму утворення та зростання оксидної плівки на поверхні металевої деталі Якщо зростання здійснюється у напрямку газового середовища, то спостерігається суттєве збільшення розміру деталі, наприклад, при оксидуванні, що необхідно враховувати в машинобудуванні. Напрямок зростання визначається співвідношенням між швидкостями процесів зустрічної дифузії іонів металу Меn+ і кисню О2 всередині оксидної плівки. Якщо швидкості дифузії іонів металу (rдиф.Mе) і кисню (rдиф.О2) сильно різняться, то зростання оксидної плівки відбувається переважно в одному напрямку, наприклад, при rдиф.О2< rдиф.Mе − в направлении газовой среды и наоборот (рис. 3, в, б). В большинстве случаев, скорости диффузии ионов металла и кислорода соизмеримы, но для ионов кислорода скорость несколько ниже из−за того, что они больше по размерам, поэтому зона роста, находящаяся в толще самой оксидной пленки, располагается ближе к ее внешней поверхности (рис. 3, а). По мере утолщения оксидной пленки процессы встречной диффузии ионов и электронов затрудняются. С повышением температуры упругость диссоциации оксидов возрастает у всех металлов. Поэтому, начиная от какой−то температуры, когда упругость диссоциации оксида металла становится больше парциального давления кислорода в воздухе (при атмосферном давлении рО2 = 0,21 ат), металл перестает окисляться и становится вполне малоактивным по отношению к кислороду. Так, например, серебро при температуре 300 ºК термодинамически еще не устойчиво, а при 400 ºК и при всех более высоких температурах упругость диссоциации Ag2O превышает парциаль14 ное давление кислорода в воздухе. При температуре 2000 ºК медь тоже становится неокисляемым металлом, однако для таких металлов, как Fе, Zn, Ni и др., даже при этих температурах упругость диссоциации окислов остается еще достаточно низкой и, следовательно, протекание реакции окисления вероятно. Необходимо учесть, что температурная зависимость для кинетики процесса окисления совершенно иная, чем для термодинамической вероятности окисления. Поэтому нет никакого противоречия в том, что термодинамическая вероятность окисления металла с повышением температуры падает, а реальная скорость окисления (при условии, что термодинамически этот процесс остается еще возможным) с повышением температуры возрастает. 2.2. Образование тонких пленок на металлах Ранее было отмечено, что в большинстве случаев образовавшиеся продукты газовой коррозии – оксиды металла остаются на поверхности металла в виде пленки. Эти пленки определяют кинетику процесса и в случае наличия защитных свойств могут привести к замедлению коррозии. Чтобы пленка оксида металла обладала защитными свойствами, она должна быть: − сплошной; − прочно сцепляться с основным металлом и обладать близким к нему коэффициентом теплового расширения; − быть химически инертной по отношению к данной агрессивной среде; − обладать твердостью и износостойкостью. Даже при обычной температуре на поверхности многих металлов и сплавов на воздухе образуются слои оксидов. Еще в 1836 г. М. Фарадей высказал предположение, что даже на блестящей поверхности металлов существует тончайшая, невидимая защитная (оксидная) пленка. Этим он объяснял пассивность железа в азотной кислоте. Если пленка пористая, рыхлая и характеризуется плохим сцеплением с основным металлом, то даже при условии инертности в данной агрессивной среде, она не будет обладать защитными свойствами. Пленки по толщине (δ) делятся на три группы: − тонкие (невидимые), δ < 4000 нм; − средние (дающие цвета побежалости), δ = от 4000 до 50000 нм; 15 − толстые (видимые), δ > 50000 нм. Товщина плівок, що утворилися при взаємодії металу із сухим повітрям, різна і залежить від роду металу, температури та інших факторів. Так, товщина плівок на меді та залозі при кімнатній температурі становить 100 – 300 нм, а на алюмінії 1000 – 1500 нм. Наведемо кілька прикладів, що доводять наявність на металах тонких плівок. 1. При нагріванні чистої блискучої поверхні пластинки заліза з одного кінця на ній з'являться інтерференційні кольори – кольори втечі. Це свідчить про появу оксидів різної товщини. Кольори йдуть у наступному порядку – від холодного кінця пластинки до гарячого, відповідно: жовтий, помаранчевий, червоний, пурпуровий, фіолетовий, синій. При випробуванні захисних властивостей утвореної плівки за допомогою крапель розчину азотнокислої міді, в місцях, де плівка має слабкі захисні властивості, відбувається реакція його із залізом: Cu(NO3)2 + Fe → Fe(NO3)2 + Cu. Внаслідок цього на поверхні під краплями з'явиться мідь. За часом, через який відбудеться реакція, можна судити про захисні властивості плівок на різних ділянках. У цьому досвіді найкращі захисні властивості виявляються на ділянці, яка розташована далеко від місця нагрівання (за жовтою зоною), де поверхня залишилася без змін. Очевидно, що на цій ділянці є дуже тонка, але щільніша плівка в порівнянні з нагрітими ділянками. 2. Якщо зламати голку (або лезо бритви) під ртуттю, то відбудеться утворення амальгами на місцях зламу. При приміщенні зламаних зразків на повітрі в ртуть амальгамування не спостерігається. Це пояснюється утворенням найтоншої оксидної плівки, що перешкоджає амальгамування. 3. При поміщенні заліза розчин гідроксиду калію з надлишком йоду лежить на поверхні заліза розчині наносять глибоку подряпину. Через деякий час залізо на місці подряпини починає розчинятися, а на дні склянки залишаються лусочки, які при подальшому дослідженні виявляються оксидом заліза (III). Процес утворення плівок на металі відбувається стадійно. 16 Перша стадія процесу взаємодії металу з корозійним середовищем – хемосорбція окислювача (O2,CO2,H2O,SO2) на поверхні металу, наприклад: Me(тв) + (O2) = Me(тв) + 2Oадс. За наявності хімічної спорідненості між металом та окислювачем реалізується друга стадія взаємодії металу з корозійним середовищем – перехід хемосорбованої плівки в оксидну. Цей процес може бути умовно описаний реакцією виду: хMe(тв) + yOадс = MexOy. Оксидна плівка, що утворюється на поверхні металу, може уповільнювати процес корозії внаслідок гальмування підведення окислювача до поверхні металу, що окислюється. У цьому випадку плівка має захисні властивості. 2.2.1. Умови суцільності плівок на металах Захисними властивостями можуть володіти лише СУСПІЛЬНІ ПЛІВКИ. Можливість утворення такої плівки визначається УМОВОМ СУСПІЛЬНОСТІ Піллінга – Бедворса. Молярний об'єм сполуки, що виникає на поверхні металу (Vok), повинен бути більшим за обсяг металу (VMe), витраченого на утворення одного моля з'єднання: Vok/VMe >1. Якщо Vok/VMe< 1, то пленка не может быть сплошной. К металлам, не удовлетворяющим условию сплошности при окислении их кислородом, относятся щелочные и щелочно−земельные металлы. Сплошность пленок – необходимый, но недостаточный фактор, определяющий ее защитные свойства. Как показал Францевич И. Н., у пленок с Vok/VMe >>1 не може бути високих захисних властивостей, через виникнення в них великої внутрішньої напруги (наприклад, MoO3 або WO3). Як верхню межу відношення обсягів приймають величину 2,5. Тоді уточнена умова суцільності виглядає: 1< Vok/VMe < 2,5. 2.2.2. Законы роста оксидных пленок на металлах Процесс роста ПОРИСТОЙ ПЛЕНКИ не осложняется процессом подвода окислителя к поверхности металла, а контролируется скоростью химической реакции образования оксида. Скорость коррозии в этом случае выражается уравнением: 17 V = dx/d = kC, где x − толщина пленки; – время; k − константа скорости химической реакции образования оксида; C − концентрация окислителя на поверхности метала. Рост пористой пленки, контролируемый скоростью химической реакции окисления металла, протекает во времени ПО ЛИНЕЙНОМУ ЗАКОНУ. Линейный закон роста имеет место: − при высокотемпературном окислении на воздухе и в среде кислорода при Vok/VMe<1; − в случае образовании летучих оксидов. Сплошные пленки затрудняют проникновение реагентов друг к другу и их рост сопровождается самоторможением процесса. Результирующая скорость этого сложного процесса определяется скоростью самой медленной стадии, т.е. возможны различные варианты контроля течения процесса. 1−й вариант. Скорость коррозии контролируется стадией массопереноса (диффузионный контроль процесса). В этом случае концентрация окислителя на границе раздела Ме – оксидная пленка равна нулю, так как скорость химической реакции весьма велика. Уравнение скорости течения коррозии определяется ПАРАБОЛИЧЕСКИМ ЗАКОНОМ роста оксидной пленки. Параболический закон роста реализуется, в частности, при окислении железа на воздухе при различных температурах. 2−ой вариант. В предыдущем случае С = 0, т.е. накопление окислителя на внутренней поверхности оксидной пленки не происходит. Если учесть возможность кинетического торможения процесса роста оксидной пленки наряду с торможением на стадии массопереноса (диффузионно−кинетический контроль процесса), в этом случае приходим к СЛОЖНО − ПАРАБОЛИЧЕСКОМУ ЗАКОНУ роста оксидной пленки. Рост оксидных пленок по сложно−параболическому закону выполняется: а) при окислении железа в парах воды или в атмосфере СО2; б) при окислении Сo, Сu в атмосфере чистого кислорода; в) при частичном разрушении оксидной пленки. Рост оксидных пленок при диффузионно−кинетическом контроле может быть выражен ТАКЖЕ СТЕПЕННЫМ ЗАКОНОМ. 18 3−й вариант. Часто рост пленки протекает медленнее, чем это следует из параболического закона роста. Это наблюдается при возникновении оксидных пленок при низких температурах (на Сu в О2 при t < 100 оС; на Аl, Тi, Ni, Zn в О2 при t = 300400 оС.) Затухание объясняется либо уплотнением пленок, либо появлением дефектов (пузырей, расслоений). В этих случаях рост толщины пленки протекает в соответствии с логарифмическим законом роста: x = ln(K). 2.3. Термодинамика процесса химической коррозии В качестве примера рассмотрим процесс окисления металла в атмосфере кислорода. Процесс окисления металла может быть представлен реакцией вида: 2Ме + nО2 → Me2n+Оn. Движущая сила процесса коррозии (ДСП) может быть определена с помощью термодинамики. Торможение процесса (ТП) не может быть определено термодинамически, однако с помощью термодинамики можно оценить условия, уменьшающие или исключающие протекание процесса (применение защитных сред, катодная защита, обескислороживание и др.). Независимо от механизма коррозии возможность ее протекания определяется знаком изменения термодинамического потенциала (см. раздел 1.1). Любое изменение энергии системы характеризует переход ее в новое состояние и является мерой стабильности системы: ΔG = GII − GI , где ΔG – энергия, израсходованная на изменение состояния системы, например, на процесс коррозии; GI − энергия системы в исходном состоянии, например, металла в конкретных условиях; G II − энергия системы в новом состоянии, например, прокорродировавшего металла. Рассчитать ΔG можно, вспомнив начало химии8. Величину изобарно-изотермического потенциала можно рассчитать по разности между суммами Δ G продуктов реакции и исходных веществ: GХР = ΔG ni – ΔG mi. (прод. р-ции) (исх. в-в). ΔG - является функцией температуры. Стандартные значения для большинства веществ сведены в справочные таблицы термодинамических величин. Изменение энергии Гиббса в зависимости от температуры учитывается уравнением Гиббса. ΔG Т = ΔН 298 – 298 Δ S 298. ΔG простого вещества = 0. Определить энергию Гиббса для любой химической реакции можно по уравнению ΔG = ΔG + RТ ln К, рассчитав предварительно величину константы равновесия, например, по равновесным концентрациям веществ. При равновесии ΔG=0, поэтому ΔG+RТlnК=0, следова8 19 2.4. Кинетика процесса химической коррозии Хотя термодинамика и дает ответ на вопрос о том, насколько изучаемая система отдалена от состояния равновесия, но ответ на весьма важный практический вопрос о скорости протекания коррозионного разрушения в рамках термодинамики получен быть не может. Рассмотрением этого вопроса и занимается кинетика коррозионных процессов. 2.4.1. Скорость процесса коррозии. Показатели химической коррозии СКОРОСТЬ КОРРОЗИОННОГО процесса может быть представлена в общем виде с помощью уравнения: СКОРОСТЬ КОРРОЗИИ (СК) = ДСП / ТП, где ДСП − движущая сила процесса; ТП − торможение процесса. Скорость химической коррозии определяют количественно по наблюдениям во времени за изменением какой−либо подходящей для этих целей величины, изменяющейся в процессе коррозии. Тогда истинная скорость коррозии металлического материала определяется из выражения: V = dy/d, где y − изменяющаяся в процессе коррозии характеристика свойств материала; − время коррозии. Для количественного выражения скорости коррозионных разрушений на практике пользуются показателями коррозии, являющимися, по сути, отражением СРЕДНЕЙ СКОРОСТИ КОРРОЗИОННОГО разрушения материала: Vср=∆y/∆. Показатели коррозии. 1. ГЛУБИННЫЙ ПОКАЗАТЕЛЬ коррозии: Kn=∆П/∆, где ∆П − глубина (средняя или максимальная) коррозионного разрушения; ∆ − промежуток времени коррозии. 2. ПОКАЗАТЕЛЬ ИЗМЕНЕНИЯ ТОЛЩИНЫ образующейся на металле пленки продуктов коррозии: Kn= ∆h/∆, где ∆h − изменение толщины образующейся на металле пленки продуктов коррозии. тельно, ΔG = – RТlnК. Данное уравнение позволяет также определить значение константы равновесия химической реакции по величине изобарно-изотермического потенциала. Для определения направления протекания многих химических реакций достаточно оценить знак Δ G. 1). При ΔН 0 , Δ S 0 реакция самопроизвольна (Δ G 0) при Т. 2). При ΔН 0, Δ S 0 реакция обратная (Δ G 0) при любой Т. 3). При ΔН 0, Δ S 0, реакция самопроизвольна (Δ G 0) при любой Т. 4). При ΔН 0, ΔS 0 реакция самопроизвольна (Δ G 0) при Т. 20 3. ПОКАЗАТЕЛЬ ИЗМЕНЕНИЯ МАССЫ Km=∆m/S∆, где ∆m − изменение массы корродирующего металла; S – площадь поверхности коррозии. 4.ОБЪЕМНЫЙ ПОКАЗАТЕЛЬ КОРРОЗИИ Kv=∆V/S∆, где ∆V − объем газа, выделившегося или поглощенного в процессе коррозии и приведенный к нормальным условиям. 5. МЕХАНИЧЕСКИЙ ПОКАЗАТЕЛЬ КОРРОЗИИ Кs=(∆s/∆)100 %, где ∆s − относительное изменение характеристики механического свойства. 6. ПОКАЗАТЕЛЬ ИЗМЕНЕНИЯ ЭЛЕКТРИЧЕСКОГО СОПРОТИВЛЕНИЯ KR=(∆R/∆)100 %, где ∆R − относительное изменение электросопротивления образца. 2.4.2. Влияние внешних факторов на химическую коррозию металлов Скорость и характер процесса химической коррозии металлов зависит от ряда факторов. ВНЕШНИМИ ФАКТОРАМИ называют те, которые связаны с составом коррозионной среды и условиями коррозии (температура, давление, скорость перемещения коррозионной среды и т.д.). Температура − мощный внешний фактор коррозии. Характер влияния температуры на скорость окисления металла определяется зависимостью константы скорости реакции окисления (k) и диффузии (D) от температуры. И k = f(T) и D = f(T) описываются одним и тем же уравнением (уравнение Аррениуса): k = k0 exp (−E/RT), (9) где k0 − константа; Е − энергия активации химической реакции. D = D0 exp (−E1/RT), (10) 1 где D0 – константа; Е − энергия активации диффузии. Таким образом, вне зависимости от вида контролирующей стадии процесса окисления с повышением температуры скорость окисления резко возрастает. Колебания температуры, особенно переменный нагрев и охлаждение, увеличивают скорость окисления металла, так как в защитной пленке образуются трещины. Состав газовой фазы. Влияние состава газовой фазы на скорость коррозии металла велико, специфично и изменяется с изменением температуры. В частности, на скорость окисления железа и 21 стали особенно сильно влияют кислород, соединения серы, водяные пары. Приведенные ниже данные свидетельствуют о зависимости увеличения относительной скорости коррозии стали с 0,17 % углерода от состава газовой фазы при 900 оС: Состав газовой фазы Кислород Чистый воздух Чистый воздух + 2 % SO2 Чистый воздух + 5 % H2O Чистый воздух + 5 % SO2+ 5 % H2O % 200 100 118 134 276 Значительное влияние на коррозию сталей и сплавов оказывают продукты горения топлива, содержащие ванадий (например, V2O5). Это соединение находится в золе от сжигания дешевого топлива − мазута, нефтепродуктов. Зола, налипая на металл, увеличивает скорость его коррозии в десятки раз. Причина этому – «ванадиевая коррозия», обусловленная легкоплавкостью V2O5, и его способностью ОФЛЮСОВЫВАТЬ (переводить в жидкое состояние) химические соединения золы и окалины. Пятиокись ванадия участвует в процессе окисления железа: 4Fe + 3V2O5 = 2Fe2O3 + 3V2O3, V2O3 + O2 = V2O5, V2O5 + Fe2O3 = 2FeVO4. Повышение содержания СО в газовой фазе понижет скорость коррозии углеродистых и низколегированных сталей, но при больших количествах СО в газовой фазе может произойти науглероживание поверхности стали. При этом возможны следующие химические реакции: 2CO + O2 = 2CO2, 2CO = Cсаж + CO2. Давление окислителя. Если бы поверхность металла не была покрыта оксидной пленкой, то скорость окисления была бы пропорциональна парциальному давлению окислителя (для двухатомных газов – PГ2). Так как поверхность металла покрыта слоем оксида, то зависимость скорости окисления от величины парциального давления окисляющего газа определяется строением защитной пленки. В то же время при окислении железа при 700 − 950о С скорость окисления не зависит от парциального давления Po2. 22 Скорость движения газовой фазы. Окисление как гетерогенный процесс определяется скоростью подвода и отвода реагентов в зону реакции. Поэтому, чем больше скорость движения потока газа, тем больше и скорость окисления металла. Режим нагрева металла. Влияние режима нагрева металла может быть рассмотрено в зависимости от влияния колебаний температуры. Иначе говоря, переменные нагрев и охлаждение увеличивают скорость окисления ввиду нарушения сохранности защитной пленки. 2.4.3. Влияние внутренних факторов на химическую коррозию металлов ВНУТРЕННИМИ называют факторы, связанные с составом, структурой, внутренними напряжениями в металле, качеством обработки поверхности металла и др. Состав сплава. Применительно к наиболее важным конструкционным сплавам на основе железа для случая газовой коррозии можно отметить следующее. 1. При высоких температурах (более 800оС) с увеличением содержания углерода в стали скорость ее окисления и обезуглероживания уменьшается вследствие более активного образования СО, что снижает окислительный потенциал газовой фазы. 2. Сера фосфор, никель, марганец практически не влияют на скорость окисления железа. 3.Титан, медь, кобальт, бериллий заметно снижают скорость окисления железа. 4. Хром, алюминий, кремний сильно замедляют окисление железа. 5. Ванадий, вольфрам и молибден могут значительно ускорить окисление стали, которое иногда носит катастрофический характер. Структура сплава. Анализ экспериментальных данных свидетельствует о том, что чем меньше в сплаве структурных составляющих, тем выше его жаростойкость. Применительно к сплавам железо – углерод наиболее устойчивой является АУСТЕНИТНАЯ СТРУКТУРА9. Аустенит (γ-фаза) − высокотемпературная гранецентрированная модификация железа и его сплавов. В углеродистых сталях аустенит − это твѐрдый раствор внедрения, в котором атомы углерода входят внутрь элементарной ячейки γ-железа во время конечной термообработки. В сталях, содержащих другие металлы (кроме железа, легированные стали), атомы металлов замещают атомы железа в кристаллической решетке, и возникает твердый раствор замещения. Фаза названа в честь сэра Уильяма Чандлера Робертс-Остина (англ. William Chandler Roberts-Austen, 1843 − 1902). 9 23 Всего различают пять больших групп нержавеющих сталей, определяемых их микроструктурой. Наиболее распространенными являются три: АУСТЕТНИТНЫЕ (AUSTENITIC) − не магнитная сталь с основными составляющими 15−20 % хрома и 5−15 % никеля, который увеличивает сопротивление коррозии. Она хорошо подвергается тепловой обработке и сварке. Обозначается буквой A в начале наименования марки. Именно аустенитная группа сталей наиболее широко используется в промышленности и в производстве элементов крепежа. МАРТЕНСИТНЫЕ (MARTENSITIC) − значительно более твердые, чем аустетнитные стали и могут быть магнитными. Они упрочняются, закалкой и отпуском подобно простым углеродистым сталям и находят применение главным образом в изготовлении столовых приборов, режущих инструментов и общем машиностроении. Больше подвержены коррозии. Обозначаются буквой С. ФЕРРИТНЫЕ (FERRITIC) стали значительно более мягкие чем мартенситные по причине малого содержания углерода. Они также обладают магнитными свойствами. Обозначаются буквой F. Стали с двухфазной АУСТЕНИТНО−ФЕРРИТНОЙ СТРУКТУРОЙ менее устойчивы против окисления. Их меньшая жаростойкость связывается с большей неоднородностью образующейся защитной пленки, что приводит к ее разрушению при росте (неоднородность возникающих внутренних напряжений). Так хромоникелевые стали с однофазной аустенитной структурой более устойчивы против окисления, чем двухфазные: Х12Н12М2Т, Х12Н9Т ведут себя лучше, чем ОХ21Н5МД2Т или 1Х21Н5Т 2]. На жаростойкость10 чугунов кроме их состава влияет и структура. Существенное влияние оказывает форма графитных включений. Так, при шаровидной форме графита стойкость против окисления выше, чем при пластинчатой. Установлено, что предварительная холодная пластичная деформация несколько ускоряет окисление металла вследствие повышения запаса его энергии. Чем тщательнее обработана поверхность металла, тем меньше скорость его окислеЖаростойкость − окалиностойкость, способность металлических материалов противостоять химическому разрушению поверхности под воздействием воздушной или иных газообразных сред при высоких температурах. 10 24 ния, что обусловлено лучшей сохранностью защитных пленок на гладкой поверхности. 2.5. Некоторые случаи газовой коррозии металлов в технологических средах В химической промышленности многие технологические процессы или их определенные стадии протекают в газовой среде в условиях повышенных температур и давлений (рис.4). При температурах от 100 до 200 − 300 °С многие газы не опасны. Химическая активность газов и скорость газовой коррозии металлов сильно возрастают при температурах выше 200 − 300 °С. Так, Cl2 начинает действовать на железные сплавы при Рис. 4. Некоторые случаи газовой температуре > 200 °С, HCl − > корозії металів у технологічних - 300 °С, SO2, NO2, пари сірих − ких середовищах. > 500 °З. Ці особливості поведінки технологічних газових середовищ та широка експлуатація їх у промисловості, вимагають докладнішого розгляду поведінки металів у реальних умовах. 2.5.1. Корозія заліза, чавуну та сталей в атмосфері О2, СО2, парів Н2О. В результаті окислення заліза за високих температур утворюється шар продуктів корозії, званий ОКАЛІНОЮ. Окалина має складну будову і включає кілька оксидів: Fe3O4 – МАГНЕТИТ має складну кристалічну решітку шпинелі; Fe2O3 – ГЕМАТИТ має ромбоедричні грати; FeO – ВЮСТИТ має дефектну структуру кристалічних ґрат. Вюстит утворюється при температурі вище 575 оС і за повільного охолодження розпадається: 4FeO Fe3O4 + Fe. 25 Нижче 575 оС вюстит в окалині відсутня і до поверхні сталі прилягає безпосередньо шар Fe3O4. Швидкість окислення сталі (див. розділ 2.4.1) з підвищенням температури зростає за законом, близьким до експонентного (рис. 5), і в координатах lgk − 1/T виражається ламаною лінією, кожен злам якої відповідає будь-якому перетворенню. Так, при температурі 575 ºС в окалині з'являється підшар вюститу FeO, що не перешкоджає дифузії кисню, в результаті чого енергія активації процесу збільшується і швидкість окислення металу збільшується. Злам кривої при температурі ~ 900 ºС відповідає алотропічному перетворенню сталі. Характер зміни температурної залежності швидкості окислення сталі вказує на те, що більш жаростійкою є аустенітна структура, при якій спостерігається повільніше зростання швидкості окислення зі збільшенням температури. Поряд з окисленням у сталях і чавуні протікає процес знеуглерожування - збіднення поверхневого шару вуглецем внаслідок взаємодії карбіду заліза, що міститься в них, з киснем і кисневмісними реагентами Fe3С+O2→3Fe+CO2 Fe3С+CO2→3Fe+2CO Fe + H2. 26 Склад газового середовища дуже впливає на швидкість окислення сталі і чавуну. При газовій корозії агресивними агентами можуть бути, наприклад, хлор, сірчисті сполуки, кисень, повітря, сполуки йоду та ін. Так, швидкість окислення Fe, С, Ni при температурі 900 °С зростає в ряді Н2О → СО2 → О2 → SО2. При цьому метали в залежності від швидкості корозії в атмосфері даних реагентів розташовуються в ряд: Ni → C → Fe. Якщо прийняти, що швидкість корозії на повітрі при 900 оС – 100 %, то добавка 2 % SO2 збільшує цю швидкість до 118 %, 5 % H2O – до 134 %, 2 % SO2 + 5 % H2O до 276 %. 2.5.2. Корозія під дією продуктів згоряння палива Продукти згоряння палива (вугілля, мазуту та ін.) у більшості випадків містять значні кількості сполук сірки (SO2, H2S) та ванадію у вигляді V2O5, який є сильним окислювачем. Легкоплавкий оксид ванадію взаємодіє із захисним шаром окалини на поверхні металу, руйнуючи його та утворюючи ванадати, які створюють з оксидом ванадію V2O5 евтектики11 з низькою температурою плавлення Fe2O3 + V2O5→2FeVO4. Так, ванадат заліза (FeVO4) має tпл = 840 оС, а температура плавлення його евтектики з V2O5 − 625 оC; температура плавлення ванадату хрому CrVO4 − 810 оС, яке евтектики з V2O5 – 665 оС. Потім ванадат бере активну участь у процесі окислення самого металу 6FeVO4+4Fe→ 5Fe2O3 + 3V2O3 4Fe+ 3V2O5→ 2Fe2O3 + 3V2O3 V2O3+O2→ V2O5, причому сам V2O5 при цьому практично не витрачається. Найбільшою мірою ванадієва корозія вражає пічні змійовики, опори та перегородки, що працюють в інтервалі температур 620 – 700 оС. Евтектика (від грец. Eutektos - легко плавиться), рідка система (розчин або розплав), що знаходиться при даному тиску в рівновазі з твердими фазами, число яких дорівнює числу компонентів системи. 11 27 При цьому швидкість корозії навіть хромистих та хромонікелевих сталей, не схильних до високотемпературної сірководневої корозії, досягає 15 мм/рік. Під дією сполук сірки залізовуглецеві сталі піддаються інтенсивній міжкристалітній корозії через більшу кількість дефектів у кристалічних решітках сульфідів, ніж оксидів. Це призводить до інтенсифікації дифузійних процесів. З підвищенням вмісту оксиду вуглецю (II) у продуктах згоряння помітно знижується швидкість газової корозії вуглецевих та низьколегованих сталей. Однак дуже висока концентрація веде до навуглерожування поверхні: 3Fe+2CO→Fe3С+CO2. Стійкими до ванадієвої корозії високолеговані сплави типу Х40Н50. Введення в сталь кремнію також позитивно впливає, легування сталі молібденом, вольфрамом, ванадієм негативно позначається на стійкості сталі, так як ці легуючі елементи промотують12 утворення V2O5. Сплави на основі Cu дуже схильні до такої корозії. Для зниження швидкості ванадієвої корозії нині використовується таке: − обмеження робочої температури до 650 оС; − використання для виготовлення вузлів, схильних до цього виду руйнування, сплавів типу Х40Н50; − вдування в паливо доломітового пилу, що містить оксиди магнію та алюмінію, які утворюють тугоплавкі сполуки з оксидом ванадію; − обмеження сумарного вмісту натрію та ванадію в паливі не більше 2 10–4 %. 2.5.3. Корозія в середовищі хлору та хлористого водню Поведінка металів у середовищі газоподібних хлору та хлористого водню принципово відрізняється від дії інших агресивних середовищ. Пов'язано це з тим, що хлористі солі, які утворюються на поверхні металу, мають низьку температуру плавлення, а в ряді випадків при підвищенні температури виганяються13, наприклад Ti + 2Cl2→TiCl4. Промотує - (синоніми) рушійний, просувний, напрямний. сублімація - Перехід речовини з твердого стану в газоподібний, минаючи рідку фазу; те саме, що сублімація. 12 13 28 Більшість таких реакцій – екзотермічні. Швидкість відведення теплоти виявляється нижчою за швидкість самої реакції, внаслідок чого метали займаються «горять» в атмосфері хлору. Це призводить до значного місцевого підвищення температури, і хлориди, що утворюються, плавляться і розкладаються. Найбільш стійкими до хлору є нікель, свинець та хромові сталі. Температура займання деяких металів в атмосфері сухого хлору: t восп. З Ti< 20 Pb 90−100 Fe 150 Cu 200 Ni > 500. У сухому хлористому водні при кімнатній температурі ряд металів і сплавів мають задовільну стійкість. З підвищенням температури стійкість металевих матеріалів поступово знижується до певної температури для кожного металу. Максимально високі температури, допустимі при тривалій роботі металів та сплавів у сухому хлорі та хлористому водні, наведені у табл. 2. Найбільш стійкими металами в сухому хлорі крім благородних металів є нікель та її сплави. Поверхневі плівки, що утворюються на нікелі та хромонікелевих сталях, мають малою летючістю та задовільними захисними властивостями. Таблиця 2 Допустимі температури при роботі деяких металів і сплавів в атмосфері хлористого водню та хлору3] Т, °С Матеріал Cl2 1200 − − 550 − 100 150 300 − Платина Золото Вольфрам Нікель Інконель (80% Fe) Мідь Вуглецева сталь Ст.З Нержавіюча сталь 12Х18Н9Т Срібло 29 HCl 1200 870 600 510-600 480 100-120 260-350 450-500 230 2.5.4. Воднева корозія сталі Воднева корозія може супроводжувати багато технологічних процесів, що протікають при підвищених температурах від 200оС і тисках від 300 МПа в середовищах, що містять водень. Ці умови відповідають таким процесам, як гідрування вугілля і нафти, синтез аміаку і метанолу та ін. Часто ці явища накладаються одне одного. Якщо в газі є аміак, то може відбуватися також і АЗОТУВАННЯ МЕТАЛУ. При контакті азотно-водневої суміші з металом в умовах підвищених температур та тиску молекулярний водень на поверхні металу дисоціює. Атомарний водень, що утворився, дифундує в решітку металу і розчиняється в ньому. При зниженні температури через зменшення розчинності водень прагне перейти в газоподібний стан усередині металу. У цьому випадку в металі виникають великі напруги, що призводять до незворотної крихкості. Воднева корозія є результатом хімічної взаємодії водню з карбідною складовою сталі. Зовнішньо прояв водневої корозії означає сильне зниження міцності сталі без помітного руйнування поверхні. Механізм водневої корозії включає наступні стадії: − при високих температурах молекулярний водень дисоціює на атоми, які сорбуються поверхнею сталі та дифундують у її кристалічні ґрати; − будучи активним відновником, атомарний водень відновлює карбід заліза і взаємодіє з розчиненим у сталі вуглецем за необоротними реакціями: Fe3C + 2H2 3Fe + CH4 C + 4H CH4, позбавляючи, таким чином, сталь її упр. Так як дифузійні процеси, у тому числі і переміщення водню, найлегше реалізуються по межах зерен, де переважно і розташовуються пластинки цементиту, їх руйнування воднем призводить до порушення зв'язку між кристаллітами і відповідно до зниження пластичності сталі. 30 Утворений метан має великий розмір молекул порівняно з параметрами кристалічної решітки фериту. В результаті цього він не може дифундувати з об'єму металу і накопичується в його мікропорожнинах і дефектах, викликаючи високий внутрішньопорожнинний тиск і призводячи до її розтріскування. При цьому тріщини розвиваються за межами зерен. У м'яких сталях з низькими властивостями міцності скупчення метану відбуваються переважно в приповерхневому шарі, утворюючи здуття, чітко виділяються на поверхні сталі. Крім участі в утворенні метану, водень має гарну розчинність у металі. Концентрація розчиненого в металі водню залежить від парціального тиску водню на межі металу − газ і може визначатися за формулою: = Ks p1/2 , де − кількість розчиненого водню в сталі, см3/100г; p – парціальний тиск водню, ат; Ks - Розчинність водню в сталі при p = 1ат. Оскільки розчинення водню металом є ендотермічним процесом, то розчинність водню збільшується з підвищенням температури. Це явище носить назву Водородопроникність (VН) 4]. Водородопроникність сталі залежить від вмісту в ній вуглецю та легуючих елементів та зменшується зі збільшенням вмісту вуглецю, про що свідчать дані, наведені в табл. 3. Таблиця 3 Вплив вмісту вуглецю на водородопроникність вуглецевих сталей Метал С, вага.% VН, см3/см2ч мм-1 Технічне залізо 0,04 5,10 Сталь 15 0,15 4,09 Сталь 30 0,27 2,79 У10А 1,10 2,24 Воднева корозія проявляється не відразу з моменту впливу водню на сталь, а через певний проміжок часу. Цей час, протягом якого не відбувається змін мікроструктури та механічних властивостей сталі, називається ІНКУБАЦІЙНИМ ПЕРІОДОМ ПРОЦЕСУ ЗНЕЗУГЛЕЖУВАННЯ сталі. 31 Інкубаційний період має важливе практичне значення, оскільки він по суті визначає безпечний час експлуатації обладнання. Інкубаційний період залежить, в основному, від температури та тиску. З підвищенням температури та тиску час інкубаційного періоду зменшується. Водночас на тривалість інкубаційного періоду великий вплив має хімічний склад сталі. Температура і парціальний тиск водню впливають як на тривалість інкубаційного періоду, а й у швидкість обезуглероживания стали при водневої корозії. 2.5.5. Карбонільна корозія Карбонільна корозія має місце у технологічних процесах, що протікають за участю вуглецю при підвищених тисках та температурах. До таких процесів відносяться, наприклад, одержання метилового та бутилового спиртів, конверсія метану та окису вуглецю. За нормальних умов СО інертна по відношенню до металів. При високих температурах і тисках оксид вуглецю вступає у реакцію з багатьма металами та утворює карбоніли. Наприклад, Fe + nCO = Fe(CO)n. Залізо з СО може утворювати три сполуки: тетракарбоніл – Fe(CO)4, пентакарбоніл – Fе(СО)5 та нанокарбоніл – Fe(CO)g. Всі ці сполуки є досить нестійкими і розкладаються при підвищенні температури. Найбільш стійке з'єднання серед них – Fe(CO)5. Утворення карбонілів збільшується із зростанням температури від 100 ºС, потім, в області температур 140 – 180 ºС спостерігається майже повна їхня дисоціація на Fe та СО. Аналогічні сполуки оксид вуглецю (II) може утворювати і з багатьма іншими металами. Карбонільна корозія викликає руйнування та розпушення поверхневого шару металу на глибину до 5 мм. Зміни структури металу на більшій відстані від поверхні вже не відбувається. 2.5.6. Корозія в неелектролітах НЕЕЛЕКТРОЛІТИ - це рідини, які не ведуть електричний струм. Рідкі корозійні середовища неорганічного походження – рідкий бром, розплавлена сірка та ін. Рідкі органічні речовини – бензол, хлороформ та ін., рідке паливо (нафта, гас, бензин та ін.), мастила. У чистому вигляді вони малоагресивні, проте, присутність у них навіть невеликих кількостей домішок (меркаптанів14, сірководню, води, кисню та ін.) різко збільшують їхню хімічну активність. Так, меркаптани і сірководень, що містяться в сирій нафті, викликають корозію Fe, Су, Ni, Ag, Pb, Sn з утворенням відповідно їх меркаптидів (Me-(S-R)n) і полісульфідів (Me2Sx). Сліди води в тетрахлориді вуглецю посилюють його корозійну активність через утворення в результаті гідролізу струмопровідних компонентів (НСl) та протікання корозії по електрохімічному механізму: CCl4 + H2O → COCl2 + 2HCl CCl4 + 2H2O + kat → CO2 + 4HCl В основному корозія протікає за хімічним механізмом. Цей процес складається з кількох стадій, кожна з яких визначає швидкість корозії: 1) дифузія окислювача до металу; 2) хемосорбція окислювача; 3) хімічна реакція; 4) десорбція продуктів корозії із поверхні металу; 5) дифузія продуктів корозії обсяг неелектроліту. У деяких випадках на металевій поверхні продуктів корозії утворюється захисна плівка, що призводить до гальмування корозійного процесу через труднощі процесу дифузії окислювача до поверхні металу. Залежно від захисних властивостей плівки, що утворюється, і її розчинності можуть встановлюватися дифузійний, кінетичний або змішаний дифузійно-кінетичний контроль процесу. Температура значною мірою впливає на швидкість процесів хімічної корозії металів у неелектролітах (див. Розділ 2.4). Вплив температури на швидкість процесу у деяких випадках ускладнюється зміною розчинності окислювача та плівки продуктів корозії металу в неелектроліті при зміні температури. 14 Меркаптани – гідросульфід похідні вуглеводнів: R – S –H. 33 Наявність води у неелектролітах значно активує корозійну дію домішок і викликає інтенсивне перебіг електрохімічної корозії металів, тобто. змінюється механізм корозійного процесу. Для захисту від хімічної корозії металів у неелектролітах підбирають стійкі в даному середовищі метали та сплави (наприклад, у крекінг – бензинах стійкі алюміній та його сплави), наносять захисні покриття (наприклад, сталь у сірководневому середовищі покривають алюмінієм). 2.6. Методи захисту металів від різних видів газової корозії 2.6.1. Методи захисту сталі від газової корозії Для захисту сталі від газової корозії найбільш широке застосування мають жаростійке ЛЕГУВАННЯ та ЗАХИСНІ ПОКРИТТЯ. Легування – (нім. legieren – «сплавляти», від лат. ligare – «зв'язувати») – додавання до складу матеріалів домішок для зміни (поліпшення) фізичних та/або хімічних властивостей основного матеріалу. Основні методи захисту металів від окислення за високих температур засновані на ЛЕГУВАННІ. Опір металів, що окислюються при високих температурах, залежить від захисних властивостей оксидної плівки, що покриває метал. Елементи, що сприяють створенню захисного шару на залізовуглецевих сплавах – хром, алюміній та кремній. Ці елементи легше окислюються при високих температурах на повітрі, ніж метал, що легується, і утворюють більш стійку окалину. Відповідно до теорії жаростійкого легування, легуючий компонент утворює на поверхні металу захисний шар, що складається тільки з оксиду легуючого компонента. Легуючий елемент повинен при цьому задовольняти наступним основним вимогам: Оксид, що утворюється на поверхні металу, повинен бути суцільним, тобто. співвідношення Vок/VMe має бути >1. Оксидний шар повинен мати високий омічний опір rМеО > rFe, розмір іонів легуючого компонента повинен бути меншим, ніж розмір іонів металу основи, тобто. rMe< rFe. Это условие вытекает из следующих соображений: 34 а) меньший радиус иона легирующего компонента по сравнению с ионом основного металла позволяет предполагать у легирующего компонента больший коэффициент диффузии в сплаве, что может обеспечить преимущественный выход ионов компонента на поверхность сплава и, следовательно, преимущественное образование оксида легирующего компонента; б) меньший радиус иона легирующего компонента позволит образовать оксид с соответствующими меньшими параметрами, который будет оказывать большое сопротивление диффузии через него и, следовательно, создавать большое затруднение для окисления основного металла. Это подтверждается сравнением размеров ионных радиусов железа и легирующих элементов: хрома, алюминия и кремния Ион металла Ионный радиус, Аº Fe2+ 0,74 Cr6+ 0,52 Al3+ 0,50 Si4+ 0,41. А энергия образования оксида легирующего компонента должна быть больше энергии образования оксида основного металла ΔGMeO<ΔGFeO. Если это условие не соблюдается, то оксид легирующего компонента будет восстанавливаться основным компонентом, как например, оксидная пленка меди, которая не может быть устойчивой на железном сплаве вследствие возможности протекания реакции CuO+Fe → FeO+Cu. При этом необходимо соблюдение условий: высокая температура плавления оксида легирующего компонента, низкая упругость диссоциации, а также отсутствие низкоплавких эвтектик в смеси с другими оксидами для того, чтобы оксид легирующего компонента был достаточно устойчив. В качестве примера можно привести элемент бор, который является аналогом Al по таблице Менделеева, но дает легкоплавкие оксиды. Температура плавления ВО3 − 294 0С, и поэтому бор, в отличие от алюминия, не может быть легирующим компонентом, повышающим жаростойкость. Необходимым условием является также, образование твердых растворов между легирующими компонентами и основным металлом, так как при этом условии возможно равномерное распределение этого элемента на поверхности и образование сплошной пленки оксида легирующего компонента. 35 Существует и другая теория, согласно которой легирующий элемент образует на поверхности сплава смешанные оксиды, обладающие повышенными защитными свойствами, по сравнению с оксидами чистых компонентов. Механизм повышения жаростойкости можно свести к тому, что легирующий компонент должен уменьшить возможность образования в окалине вюститной фазы (FeO) на стали и, таким образом, благоприятствовать образованию шпинельной фазы типа Fe3O4ּγFe2O3 с возможно меньшим параметром решетки. Жаростойкие легированные стали имеют на поверхности оксидные слои со структурой именно шпинели. Еще более высокими защитными свойствами обладают сложные шпинели типа FeOּMe2O3 или Fe2O3 ּMeO. С целью экономии легированных сталей жаростойкость углеродистых сталей повышают, применяя защитные покрытия. Для нанесения на сталь защитных покрытий пользуются следующими методами: ТЕРМОДИФФУЗИОННЫМ, НАПЛАВКОЙ И ПЛАКИРОВАНИЕМ. Для нанесения покрытий термодиффузионным методом стальное изделие после очистки поверхности механическим способом и травлением помещают в реакционную смесь, состоящую из порошка наносимого металла и катализатора. Например, при нанесении алюминиевых покрытий смесь содержит измельченный ферроалюминиевый и хлористый аммоний. Процесс насыщения поверхности стали алюминием (АЛИТИРОВАНИЕ) производится при высоких температурах (900−9500С), в результате чего происходит разложение хлористого аммония на аммиак и хлористый водород: NH4Cl → NH3 + HCl, который, в свою очередь, взаимодействует с алюминием 2Al +6HCl → 3H2 + 2AlCl3. На поверхности стального изделия происходит выделение атомарного алюминия AlCl3 +Fe → FeCl3 + Al, который диффундирует в сталь. Значительное повышение жаростойкости стали термодиффузионным покрытием обусловлено образованием окислов Al2O3 или двойных окислов FeAl2O4 в алитированном слое. На поверхности хромированных или силицированных изделий образуются соответствующие оксиды хрома или кремния. 36 НАПЛАВКА − это нанесение слоя металла или сплава на поверх- ность изделия посредством сварки плавлением. ПЛАКИРОВА́НИЕ − (фр. plaquer − накладывать, покрывать) термомеханическое покрытие − нанесение на поверхность металлических листов, плит, проволоки, труб тонкого слоя другого металла или сплава термомеханическим способом. Для защиты от карбонильной коррозии применяют хромистые стали с содержанием 30 % Сг, хромоникелевые стали с содержанием 23 % Сг и 20 % № и марганцевые бронзы доя работы при температуре до 700 °С и давлении до 35 МПа. При более низких параметрах возможно применение менее легированных сталей типа Х18Н9. Сырьем для синтеза мочевины CO(NH2)2 является NH3 и СО2. Процесс протекает при температуре 175−190°С и давлении 20 МПа. Хромистые нержавеющие стали различных марок непригодны для изготовления основных аппаратов. Наибольшую стойкость имеют стали, легированные молибденом, и хромникельмолибденовомедные стали. Важным фактором для повышения коррозионной устойчивости является тщательная очистка газов от сероводорода и дополнительное введение в систему кислорода в количестве 0,5- 1,0 об.% от содержания СО2. При сернистой коррозии сера и ее соединения - сернистый ангидрид (SO2), сероводород (H2S), меркаптаны или тиоспирты и т.д. являются достаточно агрессивными, коррозионно−активными веществами. Наиболее активным компонентом при высокотемпературной газовой коррозии является сероводород. Он даже более опасен, чем диоксид серы. Сернистый газ SO2 является исходным продуктом при производстве серной кислоты. Его получают при обжиге серного колчедана, сжигании серы, из сероводорода при утилизации отходящих газов металлургических производств. Чугунные детали скребков конверторных печей кипящего слоя, зубья и гребки колчеданных печей, котлы−утилизаторы, сухие электрофильтры, газоходы обжиговых газов в производстве серной кислоты часто выходят из строя вследствие газовой коррозии. В результате коррозии черных металлов в сернистом газе при температурах 300°С и выше образуется слоистая окалина, состоящая из FeS, FeO и др. 37 При температуре газа более 400 °С для деталей из чугуна характерно увеличение объема металла, достигающего 10 % от начальной величины. При этом резко снижается прочность материала. Детали испытывают коробление, трескаются и разрушаются. Это явление называется «ростом» чугуна и объясняется внутренним окислением металла. Максимальный рост чугуна наблюдается при 700 °С. К ростоустойчивым чугунам относятся высоколегированные хромистые чугуны, карбидный чугун типа «пирофераль» и «чугаль» Сернистый газ при высоких температурах окисляет никель. При этом образуется окалина, в состав которой входят NiS и NiO: 3Ni + S02 = NiS + 2NiO. Сернистый никель образуется и при действии на металл сероводорода: Ni + H2S = NiS + Н2. Сульфид никеля с металлическим никелем образует легкоплавкую эвтектику с температурой плавления около 625 °С. Образование этой эвтектики в сталях, содержащих никель, происходит преимущественно по границам зерен, вызывая разрушение металла. Стали с содержанием никеля выше 15 % очень чувствительны к действию сернистого газа. В процессе окисления они теряют механическую прочность. Поэтому при работе с газовой средой, содержащей диоксид серы, при температурах до 400 °С используют углеродистые стали, а при более высоких температурах − хромистые стали. Наиболее употребительны жаростойкие стали − 4Х9СА, Х6СЮ, XI7, ОХ 17 Г, X1800, Х25Т. Интенсивное образование окалины происходит при температурах выше 800−1000 °С. К жаропрочным сталям в этой среде относятся Х5М, Х6СМ, XI8Н12Т, Х23Н18. Рабочая температура для этих сплавов 550−600 °С (для Х23Н18 − 1000 °С). Сухой сернистый газ реагирует с алюминием очень медленно. Поэтому алюминий используют для защиты от коррозии деталей и узлов теплообменников и контактных аппаратов. Сухой сероводород при комнатной температуре не представляет опасности для обычных углеродистых сталей. С повышением температуры опасность сероводородной коррозии углеродистых сталей значительно увеличивается. При температуре выше 300 °С 38 железо подвергается сильной коррозии в серосодержащих газовых средах. Легирование хромом в количестве > 12% підвищує корозійну стійкість при температурі до 700-800 °С. При корозії хромистих сталей утворюється окалина, зовнішній шар якої складається із сірчистого заліза. Хром у цьому шарі практично відсутня. Весь окислений хром зосереджується у внутрішньому шарі, який і має захисну властивість. Хорошою хімічною стійкістю в атмосфері 5 10 15 20 сіроCr, мас.% водню мають феритні Мал. 6. Швидкість корозії хромисплави, що містять 25-30% стих сталей у парах нафти при 650 °С. Цифри на кривих позначають хром. зміст Н2S, % Особливу небезпеку становить спільну присутність сірчистих сполук та інших корозійно-активних компонентів. Так, у нафтовій промисловості при термічній переробці сірчистих нафт особливу небезпеку становить суміш сірководню та водню. З наведених на рис. 6 даних видно, що швидкість корозії хромистих сталей збільшується зі зростанням концентрації сірководню в парах нафти. При цьому збільшення концентрації H2S в 10 разів викликає зростання швидкості корозії більш ніж 12-15 разів. При згорянні палива утворюються складні газові суміші, що містять у своєму складі О2 та різні оксиди, у тому числі і домішки сірки. У таких випадках спостерігають СУЛЬФІДНО-ОКСИДНУ корозію. Захисна плівка на металі складається, як правило, із кількох шарів. Зовнішній шар збагачений киснем і складається з оксиду металу, а внутрішні шари, що прилягають до поверхні металу, містять підвищену кількість сірки та сульфідів. Якщо при згорянні палива утворюється зола, до складу якої входить оксид ванадію V2O5, швидкість корозії збільшується дуже швидко. Причини ванадієвої корозії сталей розібрали раніше (див. розділ 2.5.2). 39 Хромисті сталі із вмістом 4-6 % Сг вважаються напівжаростійкими. Сталі цього класу внаслідок своєї доступності, підвищеної корозійної стійкості та міцності широко застосовуються у нафтовій промисловості для виготовлення крекінг-установок. Жаростійкість цих сталей на повітрі і в топкових газах зі значним вмістом сірчистих сполук при температурах 500-600 ºС приблизно в 3 рази вище за жаростійкість нелегованих сталей. 2.6.2. Методи захисту від водневої корозії Однією з основних шляхів підвищення водородоустойчивости сталей є їх ЛЕГУВАННЯ сильними карбидообразующими елементами, що утворюють стабільніші карбіди, ніж цементит. Такими елементами є хром, вольфрам, молібден, ванадій, ніобій, титан. Великий вплив на водневитривалість сталі має тип карбідної фази. У сталях, легованих хромом, водостійкість знижується в залежності від складу карбідної фази в наступному ряду: (Cr,Fe)23C6(Cr,Fe)23C6 + (Cr,Fe)7C3(Cr,Fe)7C3 (Cr,Fe)7C3 + (Fe,Cr)3C(Fe,Cr)3C Наведений ряд показує, що найбільша водостійкість у хромистих сталях досягається при утворенні карбідів типу (Cr,Fe)23C6. Утворення цього типу карбіду при вмісті вуглецю в сталі 0,05% відбувається за наявності сталі не менше 6% хрому, а при збільшенні вмісту вуглецю необхідно підвищення вмісту хрому. При легуванні стали більш сильними карбидообразующими елементами, ніж хром, найбільша водостійкість сталі досягається при вмісті цих елементів у кількості, достатньому для зв'язування всього вуглецю в карбіди типу MeC. У випадках, коли елементи обладнання працюють і в умовах високого рівня механічних навантажень, водородостійкі сталі повинні мати жароміцність. Для цих умов застосовують сталі з 3% Cr, додатково леговані Mo, V або W, наприклад, сталь марки 20Х3МВФ (ЕІ579). Ще більшою стійкістю мають сталі марок Х5М, Х5ВФ, Х9М, 1Х13. У найважчих умовах застосовують аустетнітні жароміцні хромонікелеві сталі 0Х18Н10Т або Х17Н17М2Т. 40 Іншим широко застосовуваним способом підвищення водостійкості сталей є ПЛАКУВАННЯ їх металами, що мають нижчу водневопроникність, ніж основний метал. Це дозволяє знизити концентрацію водню на межі розділу основного металу та плакуючого шару та знизити його агресивний вплив на основний метал. Розрахунок ефективного тиску водню на межі розділу металів основа-покриття проводиться за рівнянням: 1/2 1/2 p2 = 11 (11) + + де p1 - тиск водню на зовнішній поверхні плакуючого шару; p2 – ефективний тиск водню на межі покриття-основний метал; = VН1/VН2 − відношення водородопроникності плакуючого шару та основного металу, відповідно; = l1/l2 – відношення товщини плакуючого шару та основного металу, відповідно. За зростанням водородопроникності метали і сплави можна розташувати в ряд: алюміній, мідь, нікель, Х18Н10Т, 0Х13. У практиці хімічної та нафтопереробної промисловості застосовуються переважно біметали з вуглецевих або низьколегованих сталей із захисним шаром із сталей 0Х13 або Х18Н10Т. Робочу температуру обладнання з обраної сталі слід підтримувати на 25оС нижче за ту, при якій можлива її воднева корозія. Глава 3. ЕЛЕКТРОХІМІЧНА КОРОЗІЯ МЕТАЛІВ 3.1. Електродні потенціали металів При зануренні металу в електроліт, на межі розділу фаз виникає стрибок потенціалів за рахунок утворення подвійного електричного шару 5, 6. Якщо металеву пластинку, наприклад, мідну, занурити у воду або розчин солі міді, то з шару металу, що знаходиться на межі води, позитивно заряджені іони Сu2+ почнуть переходити у воду. При цьому в кристалічній решітці металу виявляється надлишок електронів, і пластина набуває 41 негативний заряд. Між негативно зарядженою пластиною і позитивними іонами, що перейшли в розчин, виникає електростатичне тяжіння, що перешкоджає подальшому переходу іонів міді в розчин, тобто процес розчинення металу припиняється. Одночасно розвивається протилежний процес: іони міді із розчину, підійшовши до поверхні пластини, приймають від неї електрони та переходять у нейтральний стан. Через якийсь проміжок часу встановлюється стан динамічної рівноваги, при якому швидкість переходу іонів з металу розчин дорівнює швидкості розрядження іонів з розчину на металі. Схематично описане явище подано на рис. 7 (іони металу для простоти зображені негідратованими). Рівнавагу між іонами в розчині і металом для прикладу, що розглядається, виражається рівнянням: Cu2+р−р + 2ē Cuºкрист.. У рівнянні рівноваги електрохімічної реакції електрони прийнято записувати в лівій частині, тобто записувати процес відновлення. Рис.7. Схема виникнення подвійного електричного шару на межі розділу метал – розчин Як видно з наведеного вище прикладу, при контакті металу з розчином його солі, дві дотичні фази набувають протилежних зарядів. У результаті поверхні розділу фаз утворюється подвійний електричний шар і між металом і розчином виникає стрибок потенціалу (). Якщо в розчин опустити дві металеві пластини з різних металів (М1 і М11), виникне гальванічна пара, в якій відновлюватиметься менш активний метал, а окислюватиметься – більш активний (рис. 8). Мал. 8. Схема роботи гальванічного елемента У загальному вигляді робота цієї гальванічної пари визначається різницею потенціалів (ЕДС гальванічного елемента) і супроводжується балансом напівреакцій: Е = ox − red , де ox – потенціал окислювача, В; red – потенціал відновника, Ст Men+(ox) + ne ⇆ Меº Meº(red) − ne ⇄ Меn+. Коли число катіонів, що переходять в розчин в одиницю часу, дорівнюватиме числу катіонів, що осаджуються на поверхні металу, настане динамічна рівновага і процес розчинення припиниться. Тому перехід великої кількості іон - атомів металу в розчин у таких умовах неможливий. Однак у разі порушення рівноваги подвійного електричного шару шляхом розряду електронів або видалення іон-атомів металу корозійний процес протікатиме безперешкодно. Електродні потенціали металів, що у рівновазі з власними іонами, називаються рівноважними. Величина рівноважного потенціалу може бути розрахована за будь-якої активності іонів за рівнянням Нернста: E = Eº + (RT /nF)ln(aMen+), (12) n+ де Eº – стандартна різниця потенціалів при аМе =1; R – універсальна газова стала, 8,3 кДж/кмольК; T – абсолютна температура, ºК; n – валентність металу; F – число Фарадея 96 500 Кл/моль; 43 aМеn+ – активність іонів металу моль/л. Якщо підставити всі константи при 25оС (Т = 298 К) і помножити на 2,3 для переходу від натуральних логарифмів до десяткових, то отримаємо наступний вираз E = Eº + (0,0592/n)llg(aMen+), (13) При розведенні розчину потенціал металу зсувається у негативну сторону. Якщо, наприклад, активність іонів Zn2+ у розчині цинкової солі дорівнює 10–2 моль/л, то рівноважний потенціал цинку, опущеного в цей розчин, дорівнюватиме: E = − 0,76 + (0,0592/2)lg10−2 = − 0,819 В, (14) При збільшенні концентрації іонів металу у розчині потенціал металу, навпаки, зсувається у позитивну сторону. Так, якщо активність іонів цинку прийняти за 10 моль/л, то потенціал цинку буде E = − 0,76 + (0,0592/2)lg10 = − 0,73 В, (15) Електродні потенціали металів, у яких У процесі обміну, що визначає потенціал, беруть участь не тільки власні, а й інші іони та атоми, що називаються нерівноважними або незворотними. Для нерівноважних потенціалів формула Нернста непридатна, оскільки реакції, що відбуваються металі, тобто. втрата та придбання електронів, що здійснюються різними шляхами, і потенціал не може характеризувати настання рівноваги якоїсь однієї реакції на електроді. На величину нерівноважних потенціалів впливає природа електроліту, температура, рух електроліту, концентрація розчину та ін. М Na2SО4 + Н2S − 0,23 В. Через електрохімічну неоднорідність поверхні металу або електроліту на металі утворюються ділянки з різною величиною потенціалів. Основними причинами, що викликають електрохімічну неоднорідність металевої поверхні, можуть бути наступні: − ЛІКВАЦІЯ − (La liquation, Saigerung) - є здатністю сплавів розпадатися при переході з рідкого в твердий стан на складові частини або окремі сполуки, які мають різні точки плавлення; − неоднорідність захисної плівки; 44 − неоднорідність фізичних умов – неоднакова температура на різних ділянках металу, наявність деформованих ділянок, концентраторів напруги; − неоднорідність електроліту – відмінність у концентрації кисню чи концентрації солі, різна величина pH. Ділянки з негативнішим потенціалом називаються АНОДНИМИ, ділянки з більш позитивним потенціалом – КАТОДНИМИ. Ступінь гетерогенності поверхні металу визначається різницею потенціалів катодних та анодних ділянок. 3.2. Термодинаміка електрохімічної корозії Можливість мимовільного перебігу корозійного процесу визначається зменшенням вільної енергії (ізобарно-ізотермічного потенціалу) G< 0: G = −nFE < 0, (16) где n – эквивалентное число электронов; Е = К – А – эдс гальванического элемента, образующегося при контакте металла с окислителем в процессе коррозии; К – обратимый потенциал катодной реакции (окислителя); А – обратимый потенциал анодной реакции (металла – восстановителя); F – число Фарадея. Принципиальная возможность протекания процесса электрохимической коррозии металла имеет место при К > А або Е > 0. Оборотний окислювально-відновний потенціал окислювача в електроліті повинен бути в цих умовах позитивнішим, ніж оборотний потенціал металу в цих умовах. Для встановлення меж термодинамічної можливості протікання електрохімічної корозії металів можна використовувати діаграми Пурбе. Діаграми є графічними зображеннями залежності оборотних електродних потенціалів (у вольтах за водневою шкалою) від рН розчину для рівноважних станів: за участю електронів – горизонтальні лінії; за участю електронів та іонів H+ або OH– похилі лінії; за участю іонів H+ та OH–, але без участі електронів (значення рН гідратоутворення) – вертикальні лінії. 45 Рис.9. Діаграма Е - рН для системи Al-H2O при 25оС Як приклад на рис. 9 наведено діаграму Пурбе для системи Al – H2O. Кожній ділянці діаграми відповідає один термодинамічно стійкий стан: металевий стан, у вигляді іонів у розчині, у вигляді оксидів або гідроксидів. Так як встановлення рівноваги в розчині залежить не тільки від водневих, але й інших іонів, розмежування областей формується кількома лініями рівноваги. Кожна з таких ліній відповідає певній активності відповідних іонів. В області, розташованій в нижній частині діаграми, термодинамічно стійким і не схильним до корозії є металевий алюміній. У лівій частині вище горизонтальної лінії термодинамічно стійкий стан алюмінію у вигляді іонів Al3+ у розчині. Середня область відповідає стійкості твердих фаз оксиду Al2O3 або гідрооксиду Al(OH)3, що утворюють у процесі корозії на поверхні алюмінію захисну плівку. Права частина характеризує умови, за яких алюміній піддаватиметься корозії з утворенням аніону AlO2-в розчині. 3.3. Механізм електрохімічної корозії Корозія металів в електролітах відбувається при утворенні гальванічних пар, котрі називаються корозійними. Якщо опустити в посудину з розчинами хлоридів цинку та заліза, відповідно, цинкову та залізну пластини (аналогічно схе46 рН ме на рис. 8) і з'єднати їх зовнішнім провідником, то включений у ланцюг міліамперметр покаже наявність електричного струму в ланцюзі. Утворюється гальванічна пара, в якій цинк окислюватиметься і переходитиме в розчин (анод). Залізо не окислюватиметься і переходитиме в розчин, оскільки будучи з'єднане з цинком, виступає в ролі катода. Залізо, що не з'єднане з цинком, в аналогічному розчині корродирует. Насправді у складі сплавів існує велика кількість мікроелементів, різнорідних металів. Іншими словами, різнорідні метали розташовані в одній площині при безпосередньому контакті один з одним і з електролітом. В результаті цього виходить багатоелектродний елемент, в якому аноди чергуються з катодами. На анодних ділянках іони металу (Меn+) переходять у розчин. Електрони, що звільняються (nē), рухаються по металу від анодної ділянки до катодної. Розчинення іонів металу у водних розчинах слід подавати як результат взаємодії іон-атомів, що знаходяться на зовнішній поверхні металу, з полярними молекулами води. Взаємодія іонів розчиненого металу із диполями води викликається силами електростатичного тяжіння. Внаслідок цього навколо кожного іона більшою чи меншою мірою (залежно від величини його заряду) утворюється оболонка з диполів води (явище гідратації). Завдяки гідратації ефективний радіус іона ніби збільшується, внаслідок чого рухливість гідратованих іонів значно зменшується. Процес гідратації супроводжується виділенням енергії, тоді як процес дегідратації потребує витрати енергії. Крім диполь води, іон може покритися оболонкою з інших диполів. У загальному випадку це явище зветься сольватації15. Таким чином, електрохімічна корозія складається з наступних стадій, що протікають паралельно: − анодний процес, який полягає у переході іонів металу з поверхні в розчин та їх гідратації: СОЛЬВАТАЦІЯ – процес утворення асоціатів між розчинником та розчиненою речовиною, продукт сольватації − СОЛЬВАТ. Якщо розчинник – вода, то гідратація, продукт гідратації – гідрат. 15 47 Meº − nē Men+ + mH2O Men+ mH2O. гідрат - катодний процес, який полягає в асиміляції (захопленні) електронів будь-яким деполяризатором (D): D + nē . Так як анодний і катодний процеси незалежні і більш легко протікають: анодний на ділянках з більш негативним початковим потенціалом поверхні, а катодний на більш позитивних - і в практичних умовах відзначається необхідна для цього електрохімічна неоднорідність металевої поверхні, ці процеси протікають переважно локалізовано. Перебіг електричного струму здійснюється на металі - рухом електронів від анодних ділянок до катодних, а в розчині, при цьому відбувається рух іонів. Таким чином, сила струму може бути і служить критерієм швидкості електрохімічної корозії. Підрахувати матеріальну витрату анода, тобто. кількість металу, що прокородував, з 1 см2 його поверхні, можна, користуючись формулою Фарадея: К = Q A / F n = i A / F n, де К – кількість металу, що прокородував, г/см2 ; Q – кількість електрики, що протікає за час τ, [с] між анодними та катодними ділянками; i – густина струму, А/см2; F – число Фарадея; n – валентність металу; А – атомна маса металу. 3.4. Поляризація електродних процесів та її причини При замиканні електродів корозійного елемента відбувається значна зміна електродних потенціалів, а опір пари практично не змінюється. У цьому спостерігається зменшення електрорушійної сили корозійного елемента. Зміна початкових потенціалів, що призводить до зменшення корозійного струму, і, отже, швидкості корозії, називається ПОЛЯРИЗАЦІЯ. Зміна потенціалів (16) електродів у процесі роботи корозійного елемента показує, що потенціал катода стає більш негативним (КАТОДНА ПОЛЯРИЗАЦІЯ), а потенціал анода – більш позитивним (АНОДНА ПОЛЯРИЗАЦІЯ): К = К – А = А + А, (18) де К та А – значення потенціалів електродів працюючого елемента; Кº та Аo – початкові значення потенціалів електродів до замикання ланцюга; К та А – зміщення потенціалів (поляризація) катода та анода. Таким чином, різниця потенціалів E = К − А зменшується. ПОЛЯРИЗАЦІЯ є наслідком відставання електродних процесів від перетікання електронів у короткозамкненому елементі. Омічні опори (R) незначно впливають зменшення корозійного струму, оскільки вони зазвичай невеликі. Велике значення мають поляризаційні опори, які пов'язані або з утрудненнями розряду електронів на катоді (Рк), або з утрудненнями переходу позитивних іонів металу з металевих грат в розчин (РA). Ці опори, що мають омічну розмірність, є основною причиною зменшення швидкості електрохімічної корозії. Сила струму в момент замикання ланцюга відповідно до закону Ома визначається за формулою Iнач = (Кº − Аo)/R, (19). Початкове значення корозійного струму (Інач) прямо пропорційно різниці початкових значень потенціалів Кº та Аo за умови, що Кº > Аo, і обернено пропорційно опору. Корозійний струм, що встановився (Iраб.ел-та) визначається рівнянням: Iраб.ел-та = (К – А)/(R + PA + PK). Таким чином, Іраб. ел-та< Iнач. Снижению скорости электрохимической коррозии или уменьшению коррозионного тока могут способствовать: − уменьшение степени термодинамической стабильности, т. е. сближение равновесных потенциалов анодного Аº и катодного Кº процессов; − торможение катодных процессов, иначе говоря, увеличение катодной поляризуемости РК; 49 − торможение анодных процессов, то есть увеличение анодной поляризуемости РА. Поляризация является весьма желательным явлением в коррозионных процессах, так как электроды замкнуты накоротко, омическое сопротивление их невелико и, следовательно, не будь поляризационных сопротивлений, коррозия протекла бы весьма интенсивно. Всякое воздействие, уменьшающее поляризацию, увеличивает скорость коррозионных процессов. Факторы, уменьшающие поляризацию электродов коррозионного элемента, называют ДЕПОЛЯРИЗАТОРАМИ, а процессы, протекающие при этом – АНОДНОЙ И КАТОДНОЙ ДЕПОЛЯРИЗАЦИЕЙ. 3.4.1. Анодная поляризация Анодная поляризация обычно оказывает меньшее влияние на изменение разности потенциалов элемента по сравнению с катодной поляризацией. Анодный процесс, заключающийся в ионизации металла и гидратировании ионов по уравнению: Men+ + mH2O → Men+ ∙ mH2O, может протекать беспрепятственно только при условии беспрерывного отвода образующихся ионов из прианодной зоны. Торможение анодного процесса будет иметь место и в случае отставания скорости выхода ионов металла в раствор от скорости перетекания электронов на катодные участки. В результате анодной поляризации отрицательный заряд на металлической обкладке двойного слоя уменьшается, а следовательно, потенциал металла сдвинется в положительную сторону (или станет менее отрицательны). Таким образом, анодная поляризация происходит или в связи с тем, что скорость анодного процесса не поспевает за скоростью отвода электронов (перенапряжение ионизации металла), или потому, что из−за недостаточно быстрого отвода перешедших в раствор ионов металла повышается концентрация этих ионов в прианодной зоне (концентрационная поляризация). Чем легче протекает анодный процесс, тем меньше его поляризуемость. Следить за тем, легко ли протекает анодный процесс или он заметно тормозится, можно по ходу поляризационной анодной кри50 Потенциал, В вой. Эта кривая показывает зависимость потенциала электрода от плотности анодного тока на нем. Такая кривая показана на рис. 10. Удаление продуктов анодной реакции вызывает деполяризацию анода. Деполяризация анода может быть ускорена перемешиЕ Е ванием раствора, образованием о А таких продуктов коррозии, которые выпадают в осадок, а также путем образования комплексных соединений. Вследствие связывания в комплекс ионов металла, перешедших в раствор, концентрация их в прианодной зоне уменьшаПлотность тока, мА/см2 ется; анодная поляризация сниРис. 10. Анодная поляризаци- жается и переход ионов в расонная кривая твор, т.е. коррозия, усиливается. Например, медь и ее сплавы образуют в растворе аммиака хорошо растворимый комплексный ион 2+. Этим объясняется сильная коррозия меди и ее сплавов в аммиачных растворах. Поляризация анодного процесса в обычных условиях происходит в незначительной степени. Исключение представляет случай возникновения анодной пассивности. 3.4.2. Катодная поляризация Протекание катодной реакции могут тормозить два основных процесса: − торможение за счет трудности протекания самой реакции присоединения электрона деполяризатором, т. е. задержка в реакции nē + D (Dnē). Затруднение протекания катодного процесса по этой причине называется ПЕРЕНАПРЯЖЕНИЕМ РЕАКЦИИ КАТОДНОЙ ДЕПОЛЯРИЗАЦИИ; − торможение в процессе подвода к катодной поверхности деполяризатора (D) или отвода от катодной поверхности продуктов восстановления деполяризатора (Dnē). Затруднение катодного процесса по этой причине называется КОНЦЕНТРАЦИОННОЙ ПОЛЯРИЗАЦИЕЙ КАТОДА. 51 Всякий процесс захвата электрона на катоде, т.е. всякий процесс восстановления какого−либо вещества может являться катодной деполяризующей реакцией. Деполяризация катода может протекать различными путями: − ассимиляция электронов ионами: 2H+ +2ē H2, Fe3+ + ē Fe2+, Cu2+ + ē Cu+, S2O8– + ē 2SO42–; − ассимиляция электронов нейтральными молекулами: О2 + 4ē + 2Н2О 4ОН–, Cl2 + 2ē 2Cl–, Н2О2 + 2ē 2ОН–; − ассимиляция электронов нерастворимыми пленками Fe3O4 + 2ē + H2O 3FeO + 2OH–, Fe(OH)3 + ē Fe(OH)2 + OH–; − ассимиляция электронов органическими соединениями RO + 2ē + 4H+ RH2 + H2O, R + 2ē + 2H+ RH2, где R – радикал или органическая молекула. Наиболее часто встречаемыми и наиболее важными катодными деполяризационными процессами являются два: а) разряд и выделение водорода (деполяризатор ион водорода) 2H+ +2ē H2. В этом случае коррозионный процесс называется коррозией с ВОДОРОДНОЙ ДЕПОЛЯРИЗАЦИЕЙ. Этому виду коррозии подвержены электроотрицательные металлы в кислотах и весьма электроотрицательные металлы, например Mg, в воде и нейтральных растворах электролитов; б) восстановление атомов или молекул кислорода электронами, притекающими на катодных участках − КИСЛОРОДНАЯ ДЕПОЛЯРИЗАЦИЯ: О2 + Н2О + 4ē 4ОН–. Деполяризатором здесь является растворенный в электролите кислород. Этот вид коррозии является наиболее распространенным. Так корродирует большинство металлов в атмосфере, почве, нейтральных растворах электролитов. Иногда оба деполяризующие процесса протекают параллельно, как например, при коррозии железа в разбавленных растворах серной кислоты в присутствии кислорода воздуха. 52 3.4.3. Водородная деполяризация Протекание коррозии металла с водородной деполяризацией возможно, если (Me)обр < (н2)обр. (20) Согласно уравнению Нернста (н2)обр = (н2)ºобр + (RT2,3/F)lg(aH+/pH21/2), (21) о где (н2) обр – стандартный обратимый потенциал водородного электрода при всех температурах (при ан+ = 1 и рН2 = 1 ат); R – универсальная газовая постоянная. Ниже приведены значения обратимого потенциала водородного электрода (н2)обр при 25 оС и различных значениях рН среды и pН2. рн2, ат 5∙10 1 –7 0 + 0,186 0 рН среды 7 − 0,228 − 0,414 14 − 0,641 − 0,828 Изменение потенциала катода при катодной деполяризации выделением водорода подчиняется ближе всего логарифмической зависимости от плотности тока. Эта зависимость определяется выражением: К = Кº – (а + b lg i). (22) Для очень малых плотностей катодного тока (меньше 10–4 – 10–5 А/см2) потенциал от плотности тока имеет линейную зависимость. Поэтому при i = 0 К = Кº, а не бесконечности, как это следует из уравнения. Поляризуемость катода при выделении на нем водорода принято определять ВЕЛИЧИНОЙ ПЕРЕНАПРЯЖЕНИЯ ВОДОРОДА на данном материале катода. Под ПЕРЕНАПРЯЖЕНИЕМ ВОДОРОДА понимают сдвиг потенциала катода при данной плотности тока в отрицательную сторону по сравнению с потенциалом водорода в том же растворе (без наложения тока) и обозначают через η η = − (К − Кº) (23) или η = a + blgI, (24) где b – константа, не зависящая от материала, рассчитывается как 2,3 ∙ 2RT / nF; а − константа, зависит от природы и состояния поверхности материала катода и численно равна величине перенапряжения при плотности тока, равной единице. 53 Поляризационная кривая для случая водородной деполяризации имеет вид кривой Кº MN (рис. 11). − N 1 M η Рис. 11. Катодная поляризационная кривая для процесса водородной деполяризации ко или (н2)обр i1 0 i P Если по оси абсцисс взять не плотность тока, а логарифм этой величины, то кривая перенапряжения будет прямой. В этом случае константа b является тангенсом угла наклона полученной прямой к оси абсцисс, константа а равна величине ординаты lg i = 0 (т.е. при плотности тока i = 1). Величина перенапряжения водорода зависит от плотности тока, т.е. при данной силе тока, от истинных размеров поверхности катода. При одинаковой силе тока она больше на гладкой поверхности, чем на шероховатой. 3.4.4. Кислородная деполяризация Процессы коррозии с КИСЛОРОДНОЙ ДЕПОЛЯРИЗАЦИЕЙ могут протекать в том случае, если при данных условиях обратимый потенциал металла отрицательнее обратимого потенциала кислородного электрода (Me)обр < (О2)обр. (25) 54 Обратимый потенциал кислородного электрода может быть вычислен по уравнению Нернста: (О2)обр = (О2)ºобр + (RT2,3/4F)lg(pО2/a4ОH−) (26) где (О2)ºобр – стандартный обратимый потенциал кислородного электрода (при аOH– = 1 и рО2 = 1 ат); рО2 – парциальное давление кислорода; аОН– – активность гидроксильных ионов. Величина обратимого потенциала кислородного электрода (О2)ºобр при 25 oC для различных значений рН и рО2: рН среды ро2 , ат 0,21 1 0 + 1,218 + 1,229 7 + 0,805 + 0,815 14 + 0,391 + 0,400 Коррозия с кислородной деполяризацией является термодинамически более возможным процессом, чем коррозия с водородной деполяризацией, так как равновесный потенциал восстановления кислорода более положителен, чем равновесный потенциал выделения водорода. Коррозия металлов с кислородной деполяризацией является самым распространенным коррозионным процессом. Коррозии с кислородной деполяризацией подвергаются металлическая обшивка нефтезаводской аппаратуры и оборудования, детали аппаратов, соприкасающиеся с водой и нейтральными водными растворами солей (например, трубы конденсационно−холодильного оборудования), подземные трубопроводы, днища резервуаров и др. При коррозии металлов с кислородной деполяризацией катодный процесс определяется перенапряжением катодной реакции и скоростью подвода кислорода к электроду. Замедленность катодной реакции по схеме: О2 + 2Н2О + 4е 4ОН– (в щелочных или нейтральных растворах), или по схеме: О2 + 4Н+ + 4е 2Н2О (в слабо−кислых растворах) называется ПЕРЕНАПРЯЖЕНИЕМ ИОНИЗАЦИИ КИСЛОРОДА. При больших плотностях тока (при iк > 10–2 А/м2) та значній швидкості підведення кисню до катода перенапруга іонізації кисню має логарифмічну залежність від щільності струму 55 δ" = а"+ b" lg i, (27) де δ" − перенапруга іонізації кисню; a" − константа, яка залежить від матеріалу катода та стану його поверхні, температури та інших факторів. Вона чисельно визначається як величина перенапруги при i = 1; b" − константа, яка не залежить від матеріалу катода; вона визначається з рівності b" = 2RT/nF ∙ 2,3; при температурі 20о і n = 1, b" = 0,117. Механізм катодного відновлення кисню досить складний і протікає кілька стадій: розчинення кисню в електроліті, перенесення розчиненого кисню до катодним ділянкам металу, іонізація кисню. На відміну від водневої деполяризації, на яку концентраційна поляризація практично несуттєва, при корозії металів з кисневою деполяризацією вона грає дуже велику роль внаслідок обмеженості швидкості підведення кисню до катода. Це пояснюється невеликою розчинністю кисню в розчині електроліту і, отже, його малою концентрацією, складністю дифузії кисню через нерухомий шар рідини, що прилягає до катода. При сильному перемішуванні розчину, інтенсивної циркуляції або умовах, що забезпечують значну аерацію електроліту, корозійний процес гальмується перенапругою іонізації кисню. Дифузійні процеси позначаються на швидкості корозії при зануренні металу в спокійний або в розчин, що злегка перемішується. Загальна крива катодної поляризації має складний вигляд (рис. 12) і є сумарною із трьох кривих, що характеризують поляризацію при іонізації кисню (AB), концентраційну поляризацію (DE) та розряд іонів водню (GFH). У першому ділянці швидкість катодного процесу, тобто. кількість кисню, що відновлюється, в одиницю часу, менше максимально можливої швидкості доставки кисню до поверхні шляхом дифузії. У зв'язку з цим у цій ділянці швидкість процесу обмежується переважно уповільненістю процесу відновлення кисню. Рис.12 Катодна поляризаційна крива для процесу кисневої деполяризації При подальшому підвищенні щільності струму потенціал зміщується в негативному напрямку спочатку поступово, а потім стрибкоподібно (у). Різке усунення потенціалу в негативному напрямку пов'язане з концентраційною поляризацією внаслідок зменшення концентрації кисню. При деякому значенні потенціалу стає можливим новий катодний процес, який зазвичай пов'язаний з водневою деполяризацією (ділянка EK). 3.5. Контролюючий фактор корозії Процес електрохімічної корозії металів та сплавів складається з елементарних процесів чи стадій. Реальна швидкість корозійного процесу знаходиться у прямій залежності від сумарного гальмування його на кожній стадії. Частка гальмування загального процесу кожної з його стадій характеризує СТУПЕНЬ КОНТРОЛЮ ПРОЦЕСУ даної стадією. 57 називають таку стадію, яка визначає швидкість корозії, оскільки має найбільший опір у порівнянні з рештою стадій. КОНТРОЛЮЮЧИМ ПРОЦЕСОМ А (Me)обр Q Q N M (Me)обр о2)обр imax I imax б б а (2)обр в А А imax (Me)обр (Me)обр K ∆К R P (2) I I S (2)обр i I р р д д Рис. 13. Поляризаційні корозійні діаграми для основних практичних випадків контролю електрохімічних корозійних процесів 58 I У практичних умовах мають місце кілька основних випадків контролю електрохімічної корозії металів і сплавів, обґрунтованих Н.Д. Томашовим. Беккерєв І. В. «Метали та сплави – Марки, хімічний склад», Частина 2, Ульяновськ: УлГТУ, 2007. − с. 630, http://www.bibliotekar.ru/spravochnik-73/index.htm. 3]. Семенова І.В. Корозія та захист від корозії, М.: Фізматліт, 2002, 335 с. 4]. Халдєєв Г. В., Борисова Т. Ф. «Воднепроникність металів і сплавів у корозійно-електрохімічних процесах. Електрохімія» - Том 30, Москва, 1989 с. 232 5]. Т. Л. Луканіна, Т. Т. Овчиннікова, В. Я. Сігаєв, "Загальна хімія" II частина. Навчальний посібник. 2 – е видання, виправлене та доповнене, 2006 р.)